Properties Characterization and Identification of Saponarin and Ribosnarin (apigenin-6-C-ribosyl -7-O-glucoside) Extracted Abundantly from the White-Color Moth Orchid Flowers (Phalaenopsis Hybrids)
by Po-Chang Chiu1, Yu-Jang Li2, Chih-Yu Lo1, Shu-Mei Lin1, Chenwei Huang1, Robin Y.-Y. Chiou1*
1Department of Food Science, National Chiayi University, Chiayi 60004, Taiwan, ROC.
2Department of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan, ROC.
*Corresponding Author: Dr. Robin Y.-Y. Chiou, Department of Food Science, National Chiayi University, Chiayi 60054, Taiwan.
Received Date: 27 September 2024
Accepted Date: 4 October 2024
Published Date: 07 October 2024
Citation: Chiu PC, Li YJ, Lo CY, Lin SM, Huang C, Chiou RYY (2024) Properties Characterization and Identification of Saponarin and Ribosnarin (apigenin-6-C-ribosyl -7-O-glucoside) Extracted Abundantly from the White-Color Moth Orchid Flowers (Phalaenopsis Hybrids). Food Nutr J 9: 314. https://doi.org/10.29011/2575-7091.100314
Abstract
In addition to ornamental purposes of the moth orchid flowers (Phalaelopsis hybrids), flower-bearing C-glycosylflavonoids and properties characterization deserve investigations. In this study, the petals and sepals of 9 different-colored moth orchid flowers were extracted with 60% methanol for HPLC analysis. Two noticed peaks were detected and varied with cultivar. Addressed on the flower components of white-color Phalaenopsis (Sogo Yukidian), the peak areas from lips were lower than those of petals or sepals. When the components were removed either from fresh or wilted flowers and dried at 60 oC prior to pulverization, extraction and HPLC analysis, both peak areas in each components varied limitedly. After purification of the flower extracts by medium-pressure liquid chromatography and semi-HPLC fractionation, and elucidation by NMR and ESI-MS analyses, apigenin6-C-glucosyl- (saponarin) and -6-C-ribosyl-7-O-glucopyranoside (ribosnarin) were identified. Both are C-glycosylflavonids and used as authentic references in HPLC quantification of saponarin and ribosnarin in 11 collected white-color moth flowers, contents were 1.5±0.5 and 5.6±1.1% (dry basis). In ABTS∙⁺ scavenging and tyrosinase activities determination, both compounds exhibited ABTS∙⁺ scavenging activities with dose dependency and no obvious tyrosinase inhibitory activity in the test concentrations from 0 to 100 µM. In substrate availability determination, both compounds were equally mixed with nitrophenyl-β-D-glucopyranoside (pNPG) or each one was alone mixed with pNPG and subjected to incubation with almond β-glucosidase at 60 oC for 10 min, only pNPG was hydrolyzed, indicating that both are not β-glucosidase hydrolysable. As generalized, the assessed activities were not differentiated by 6-C-gluco- and 6-C-ribo- molecularly different moiety. Since both exist abundantly in either fresh or wilted flowers, it is worthy to use the flowers as a potent source of C-glycosylflavonoids to explore their potencies for the known and unknown promising products development.
Keywords: Phalaenopsis Hybrids; Moth Orchid; C-Glycosylflavonoids; Saponarin; Ribosnarin (Apigenin-6-Cribosyl-7-O-glucopyranoside); β-Glucosidase
Introduction
Plant-biosynthesized C-glycosylflavonoids are widespread in nature and attract research interest mainly due to their C-C sugar linkage which is different from that of the corresponding O-glycosides. Under normal physiological condition or acid condition, C-glycosides are not cleaved and bear better bioavailability than the O-glycosides, thus, rendering a wide spectrum of difference in biological and pharmacological properties [1,2]. In comparison to their O-linked counterparts, C-glycosylflavonoids are comparatively less presence in nature and their distribution, quantity, and biological effects in the human diet have received little attention. Therefore, their unique structural and activity features, such as the di-C-glycosylflavones, have motivated organic chemists to develop synthetic approaches with an attempt to mass production of such bioactive molecules [3,4]. Along with the current trend indicating increased researches and efforts have been contributed on production and potential applications of C-glycosylflavonoids, further investigations in both discovery and properties characterization of such novel natural sources are equally of importance.
For the moth orchids (Phalaelopsis hybrids) with various colors, shapes, flower numbers and flagrances for gardening and ornamental purposes which are popularly cultivated in Taiwan mainly for domestic and overseas markets. In the past, most attentions have been focused on the flower characteristics during the blossom seasons. In the recent years, moth orchids including leaves, roots, and flowers destined as a source of bioactive phytochemicals including anthocyanins, phenolic compounds and diverse flavonoids have attracted research attention. These studies are conducted with an attempt to explore additional profits achieved from development of the value-added products [5-9]. As investigated [6,9], a new C-glycosylflavone, i.e., apigenin-6-Cribosyl-7-O-glucoside along with a known saponarin (apigenin-6C-glucosyl-7-O-glucoside) have been isolated and identified in the red-color flowers of Phalaenopsis hybrids (Dtps. Tinny Ribbon × Dtps. Plum Rose). Both are di-C-glycosyl-O-glycosyl flavones and molecularly differ by a C-linked sugar moiety. Both saponarin extracted from the barley young leaves and isosaponarin extracted from the wasabi leaves are di-C-glycosyl-O-glycosyl flavones which have been intensively investigated to highlight their specific bioactivities [10-13]. In this study, based on the fact that moth orchids bloom in a limited period and most of the wilted flowers are discarded, the out-season or senescent flowers may provide a low-cost source for production of the bioactive phytochemicals for related value-added products development. Investigations were accordingly addressed on extraction and HPLC analysis of the petals, sepals and lips of 9 different-colored moth orchid flowers, two noticed peaks (I and II) have been detected and varied dependently on cultivar. Peak areas of the extracts from the fresh or wilted flowers after subjection to artificial drying were compared. In addition to extraction, purification and identification, further biological activities assessments including ABTS∙⁺ scavenging and tyrosinase inhibitory activities and substrate availability for ß-glucosidase hydrolysis caused by the specified molecular difference were extended.
Materials and Methods
Moth Orchid Flowers (Phalaenopsis spp.)
In this study, blossoms of Phalaenopsis Sogo Yukidian (Figure 1C) along with other 8 Phalaenopsis hybrids (moth orchid) in pods with different colors (Figure 1) were provided by Ai-Lan Flower Studio (Chiayi, Taiwan). Except specifically described, the flower components including 2 petals, 3 sepals and 1 lip from each moth flower were used as raw material.
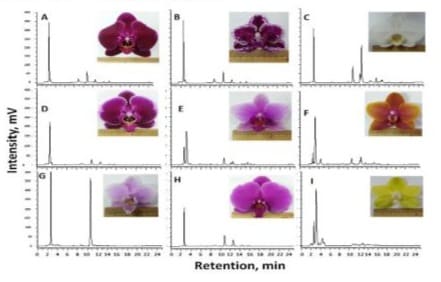
Figure 1: Different-colored orchid flowers subjected to 60% methanol extraction and HPLC analysis monitored at 254 nm; A: Phalaenopsis Fusheng Black Pearl; B: Phalaenopsis Yu Pin Burgundy; C: Phalaenopsis Sogo Yukidian; D: Phalaenopsis Fusheng Pink Pearl; E: Phalaenopsis Kano Beauty; F: Phalaenopsis Sogo Lawrence; G: Phalaenopsis Jia Ho Pink Girl; H: Phalaenopsis Fusheng Purple Gem; I: Phalaenopsis Sogo Medal.
HPLC Analysis of the Flower Extracts with Different Colors
From each of the fresh flowers with various colors as shown in Figure 1, the flower components (2 petals, 3 sepals and 1 lip) were manually cut with a pair of scissors, the components were respectively pooled as sublots and weighed. Components in each sublot were chopped into small pieces and deposited into a 100 mL flask and homogenized with 10 volumes (v/w) of 60% methanol with a polytron homogenizer (Polytron 3100D, Kinematica AG, Luzern, Switzerland) rotated at 15,000 rpm for 2 min and ultrasonicated for 10 min. Then, the homogenate was centrifuged (9,000 g) for 3 min. The supernatant was membrane-filtered (0.45 μm) for HPLC analysis (Hitachi L-7100 pump, L-7455 PDA detector; Hitachi Co., Ltd., Tokyo, Japan) equipped with a RP-18 column (Hypersil ODS column, 250 x 4.6 mm, Thermal Hypersil Ltd., Chesire, UK). The injection volume, flow rate and monitoring wavelength were 20 μL, 1 mL/min and 254 nm, respectively. The mobile phase comprised methanol (A) and water (B) and run by a programmed dual solvent system set as 0 min with A 20%; 15 min with A: 60%; 20 min with A 100%; 25 min with A 20%. As primarily observed, peaks with ca. 10.5 and 11.5 min of retention time have been detected in all cultivars of flowers even the yellow-colored one (Figure 1I). In comparison, both peaks with fairly high peak areas were detected in the flowers of Phalaenopsis Sogo Yukidian (Figure 1C). Thus, the whitecolored flowers were selected as targets for the two noticed peak substances and subjected to the followed extraction, isolation and structural identification.
Peak I and Peak II Distribution in Flower Components as Affected by Drying
Freshly harvested white-color flower components of petals, sepals and lips were separated, pooled and further divided into three sublots. From each sublot after chopped into small pieces were weighed and dried a forced-air oven at 60 oC for 18 h. Dry solid contents for three flower components was ca. 12.5% (wet basis). Based on the resulted dry solid contents, the original moisture contents were estimated and equivalent to ca. 87.5% (wet basis). Accordingly, in order to extract from the same dry solid basis, 10 volumes (v/w) 60% methanol was applied for extraction for the fresh flower components, while 80 volumes (v/w) 60% methanol was applied for extraction from the dried flower components.
Compositional Variation of the Naturally Wilting Flowers
From the naturally wilted white moth flowers, the petals, sepals and lips were separated, pooled and divided into three sublots. Each sublot was subjected to drying at 60 oC for 18 h and pulverized into powder. From which 0.5 g was sampled and extracted with 40 mL (80/1, v/w) of 60% methanol for extraction and HPLC analysis following the procedure described above.
Extraction and Isolation of the Targeted Compounds
As done preliminarily with other 10 collected white color moth flowers from flower markets along with Phlaenopsis Sogo Yukidian, their HPLC chromatograms varied limitedly. For mass extraction, all collected petals and sepals were pooled for drying at 60 oC overnight for 18 h and pulverized into powder and stored at – 20 oC in a sealed bottle for later use. With an attempt to save solvent used, 10 g powder was homogenized with 100 mL of 60% methanol solution (1/10, w/v) for 2 min (15,000 rpm, Polytron PT3100D) and ultra-sonication for 10 min. The homogenate was centrifuged at 9,000 g for 10 min at 20 oC (Hitachi Himac Centrifuge SCR 20B, Hitachi Co., Tokyo, Japan). After membrane filtration (0.45 μm) of the supernatant, the filtrate was diluted with 3 volumes of deionized water and loaded onto a solid-phase extraction (SPE) cartridge (C18 10 g-cartridge, Supelco Inc., Bellefone, PA) and eluted with deionized water and 25% methanol solution for cleaning-up. Then, the column was eluted with 15 mL of 80% methanol and the eluent was vacuum evaporated to dryness. The residue was dissolved in appropriate volume of 60% methanol and membrane-filtered (0.45 µm) for medium pressure liquid chromatographic fractionation (MPLC) (Buchi Pump Manager C-615 with a Pump Module C-601, Buchi Labortechnik, Flawil, Switzerland).
For MPLC, an open column (ODS, 3.6 × 20 cm of bed volume, Buchi Glass Column) was run with an isocratic solvent of 70% methanol set at 20 mL/min of flow rate. The eluent was tubecollected every 1 min and each was subjected to analytical HPLC analysis as that described above. The tubes containing the targeted compounds were collected, pooled and concentrated by vacuum evaporation by Buchi Multivapor (P-6) connected and controlled with a Vacuum pump (V-700) (Buchi Labortechnik, Flawil, Switzerland). The concentrate was membrane filtered (0.45 μm) and subjected to semi-HPLC (Hypersil ODS column, 250 x 10 mm, Thermal Hypersil Ltd., Chesire, UK) run with 3 mL/min of flow rate and 100 μL of injection volume. Each run was programmed with a dual-solvent system comprising A: methanol and B: water set at 0 min: A 40%; 15 min: A 50%; 20 min: A 40%; 25 min: A 40% monitoring at 254 nm. The eluents under each of the two targeted peaks were respectively collected, pooled and vacuum evaporated to dryness. The dried substances were further dried in a chamber under vacuum at 40 oC overnight for weight determination. The residues were cap-sealed for storage under – 20 oC for structural identification and bioactivity assessment. As obtained 4.2 and 7.6 mg of the peak I and II compounds were recovered.
NMR Spectroscopic Analyses
For NMR analyses, d-methanol was used as the solvent to dissolve the purified compounds for NMR 1D and 2D analyses. The samples were subjected to 1H NMR (Bruker Avance III 500 MHz NMR spectrometer, Bruker BioSpin GmbH, Rheinstetten, Germany) and 13C NMR analyses equipped with a 5-mm broad BBFO probe and Topspin 2.1 software. The 1H and 13C NMR were measured at 298 K operating at 500.13 and 125.75 MHz, respectively. The spectra were recorded in d-methanol at 298 K. The 13C spectrum was accumulated by running for 20,000 scans. The chemical shifts were recorded with reference to the residual solvent signal at 3.35 and 4.70 ppm for d-methanol) and coupling constants (J) in Hertz (Hz). The 13C-DEPT and COSY analysis of orchid peak II for d-methanol solvent were extended. All 1D (1H and 13C) and 2D NMR measurements were performed with standard Bruker pulse sequences.
LC-ESI-MS Analysis
A Thermo LC-ESI-MS system equipped with a syringe pump, and an LXQ linear ion trap mass detector (Thermo Finnigan, San Jose, CA) incorporated with an electrospray ionization (ESI) interface was used for MS analysis. The acquisition parameters were set as follows: the negative ion polarity mode was selected for the ESI ion source with the voltage on the ESI interface maintained 3.2 kV; nitrogen gas was used in sheath gas at a flow rate of 5 arbitrary (arb) units and auxiliary gas at a flow rate of 0.1 arb units; capillary temperature, 200 °C; capillary voltage, -5 V; tube lens offset voltage, -70 V; and mass range, from m/z 300 to 700. In tandem mass spectrometry, the base peak ion was monitored with a relative collision energy setting at 25. The MS system was controlled by Xcalibur 2.06 software. Two samples were respectively dissolved in methanol for flow injection analyses.
Peak I and peak II Contents in White-Color Moth Flowers
In addition to Phalaenopsis Sogo Yukidin, another 10 white color moth flowers were collected from the local gardens or florist shops. The petals and sepals were removed and dried at 60oC overnight for 18 h, then pulverized into powder and subjected to extraction and HPLC analysis following the above described procedure. The previously purified peak I and II as authentic compounds were used to prepare a series of concentrations in 60% methanol solution and subjected to HPLC analysis to construct standard curves. Based on the curves, peak I and II concentrations in the dried flower powders from various sources were estimated on dry solid basis.
ABTS∙⁺ scavenging and Tyrosinase Inhibitory Activities Determination
For ABTS∙⁺ scavenging activity determination, the procedure of Lin et al. [14] was followed with minor modification. Briefly, 5 mL of 14 mM ABTS [2,2’-azino-bis (3-ethyl-benzthiazoline-6-sulfonic acid)] solution was mixed with 5 mL 4.9 mM potassium persulfate and reacted under dark condition for 12 to 16 h for formation of the dark blue ABTS∙⁺ stock solution. Prior to examination, the stock solution was diluted with pure water to 0.70 ± 0.05 with absorbance at 734 nm. From this, 1.95 mL was withdrawn and mixed with 50 μL of test sample solution dissolved in 20% DMSO and reacted at the ambient temperature under dark condition for 6 min, followed by absorbance determination at 734 nm. A series of orchid peak I, peak II and Trolox (as a control) solutions were respectively dissolved in 20% DMSO to prepare 0, 12.5, 25, 50 and 100 μM and run determinations concurrently to measure absorbance at 734 nm. The activities were directly expressed by absorbance at 734 nm, indicating that the lower absorbance is the higher scavenging activity.
For tyrosinase activity determination, the purified peak I and peak II compounds were dissolved in 20% DMSO for preparation of a series of real reaction concentrations at 0, 12.5, 25, 50 and 100 µM (prepared stock concentrations at 0, 37.5, 75, 150 and 300 µM) solutions. Authentic DOPA (3,4-dihydroxyl L-phenylalanine) (D 9628, Sigma-Alderich Co., St. Louis, MO) dissolved in potassium phosphate buffer (0.05 M, pH 6.7) was freshly prepared and used as enzyme substrate. Mushroom tyrosinase (Sigma T3624) dissolved in the phosphate buffer containing 200 units/mL was used as enzyme solution. For each determination, 0.3 mL substrate and 0.3 mL peak I or peak II solution were pre-mixed and incubated at 30 oC for 10 min. Then, 0.3 mL tyrosinase solution was introduced, vortexed and deposited into a quartz cuvette for continuous absorbance measurement at 490 nm for 60 sec by running a time scan mode with a spectrometer (Hitachi U-5100, Hitachi Co.). For each test concentration, absorbance at 0, 20 30 and 60 sec were selected for regression to construct a linear equation and based in determination of slope and correlation coefficient (R2). A series of koji acid (Sigma K-3125) solutions in 20% DMSO were run concurrently as a positive reference. A blank was run by a reaction containing DOPA substrate, tyrosinase and 20% DMSO in the cuvette and absorbance at 490 nm was continuously measured. For each test compound at various concentrations, linear equations with slopes were determined and tyrosinase inhibitory activity was expressed by decrease of slope as affected by increase of concentration.
Substrate Availability Assessment for β-glucosidase hydrolysis
The procedure of Silva et al. [15] was followed with modification. As a preliminary experiment to check enzyme activity, almond β-glucosidase (EC 3.2.1.21, Sigma G-0390) 96 mg was weighed and dissolved in 2.4 mL sodium acetate buffer (0.05 M, pH 5.0) containing ca. 100 enzyme units per mL. For substrate preparation, 1,200 µM p-nitropheyl-β-D-glucopyranoside (pNPG, Sigma N7006) was prepared in 20% DMSO. For each enzyme reaction, 100 µL pNPG was mixed with 100 µL 20% DMSO, 0.5 mL β-glucosidase solution and incubated at 60oC for 10 min. Then, 100 µL of the reactant was withdrawn and heated in boiling water bath for 5 min prior to cooling to the ambient temperature. As a control, 0.5 mL of β-glucosidase solution was inactivated by heating in boiling water bath and, after cooling to ambient temperature, was introduced to mix with 100 µL pNPG and 100 µL 20% DMSO for incubation at 60 oC for 10 min. And from which 100 µL was withdrawn and heated in boiling water bath for 5 min prior to cooling to the ambient temperature for the following HPLC analysis.
For enzymatic activity determination by HPLC analysis, the reacted solution was centrifuged at 9,000 g for 3 min and the supernatants were subjected to HPLC analysis following the same gradient dual solvent running program described above for analysis of orchid peak I and peak II. As observed, pNPG and the nitro-phenol released by hydrolysis of β-glucosidase could be HPLC detected by monitoring at 254 nm (Figures 7A1 and 7A2).
In substrate availability assessment, each 600 µM of the purified orchid peak I and II was respectively prepared in 20% DMSO as a stock solution. From which 100 µL peak I was mixed with 100 µL peak II and further mixed with 0.5 mL β-glucosidase solution for incubation at 60 oC for 10 min. Then, from which 100 µL was withdrawn and heated in boiling water bath for 5 min and followed by HPLC analysis described above for enzymatic hydrolysis determination. For respective substrate availability assessment, 100 µL peak I was mixed with 100 µL 1,200 µM pNPG or 100 µL peak II was mixed with 100 µL 1,200 µM pNPG. Then, each was respectively introduced with 0.5 mL β-glucosidase solution for incubation at 60 oC for 10 min. Similarly, from each after reaction, 100 µL was withdrawn and heated in boiling water bath for 5 min and following the HPLC procedure described above for enzymatic hydrolysis determination.
Statistics
Triplicate determinations were conducted and means with standard deviation are presented.
Results and Discussion
Moth Orchid Cultivars with Different Colors and HPLC Analysis of the Extracts
In this study, 9 different-colored moth orchid flowers (Figure 1) were collected and from each the petals combined with sepals were subjected to 60% methanol extraction and HPLC analysis. Based on the HPLC chromatograms, two peaks with ca. 10.5 and 11.5 min of retention time have been detected in all test flowers and deviated remarkably with flower colors. These two peaks were designated as orchid peak I and II for the following specification. As noticed, peak I was dominantly detected in the flowers of Phalaenopsis Jia Ho Pink Girl (Figure 1G). For the yellow-colored flowers of Phalaenopsis Sogo Medal (Figure 1I), both peaks were detected in much lower quantities than others. It was further observed that both peaks were detected with fairly high quantities in the white-color Phalaenopsis Sogo Yukidian flowers (Figure 1C). The white-color moth orchid is originated from Taiwan and historically named Phalaenopsis taiwanambilis (meaning grandmother of the white moth orchids) which are popularly cultivated in Taiwan for ornamental purpose. Thus, the flowers were selected in this study as a convenient source in providing sufficient raw material to facilitate the followed extraction, purification and properties characterization.
HPLC Analysis of the Extracts of Flower Components as Affected by Drying
With an attempt to compare contents of the two peak substances distributing in the flower components, sublots of 3 sepals, 2 petals and 1 lip removed and pooled from each of the flowers were respectively extracted for HPLC analysis. Two peaks were detected in all test extracts and the peak areas of lips (Figure 2C1) were comparatively lower than those of sepals and petal (Figures 2A1 and 2B1). For further comparison of the peak areas as affected by wilting (senescent) or artificial drying, the fresh and wilted flower parts were respectively separated and pooled for drying at 60 oC for 18 h. Each of the sublots after drying was pulverized into powder and subjected to extraction and HPLC analysis (Figure 2). For comparison of the peak I and II areas for each of the flower component counterparts, there was very limited difference in either of the petals, sepals or lips. However, in regardless of the noticed two peaks, there are more polar fractions with retention times 2 to 5 min were detected in the fresh and fresh-dried counterparts (Figures 2A and 2B) than in the wilt-dried counterparts (Figure 2C). Presence of these fractions in relation to flower senescence remains research interest. In addition, one detected peak with retention time ca. 23.5 min was detected in the fresh flowers after subjected to artificial drying (Figures A2, B2 and C2). In addition to the two noticed peaks, these observed compositional alternations would provide optional approaches in raw material collection and treatment to optimize the related processing.
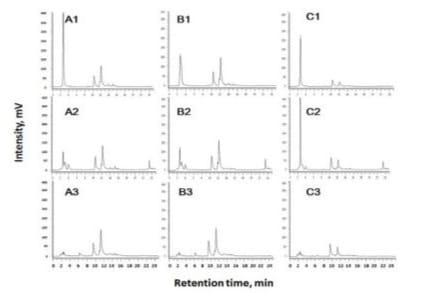
Figure 2: HPLC chromatograms of the sepals (A), petals (B) and lips (C) of the white color Phalaenopsis flowers subjected to 60% methanol extraction and followed by HPLC analysis monitored at 254 nm; 1: freshly harvested flowers; 2: fresh flowers after drying at 60 oC overnight (18 h); 3: wilted flowers after drying at 60 oC overnight (18 h).
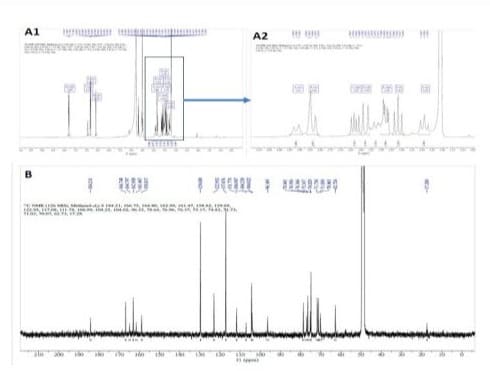
Figure 3: 1H and 13C NMR spectroscopic spectra of orchid peak II: A1: full 1H NMR spectrum; A2: extended 1H spectrum addressed on the sugar signals; B: 13C NMR spectrum.
Peak I and II Purification, Identification and Used for Quantification in 11 Collected White Color Moth Orchid Flowers
From the 60 oC-dried flower powder, 10 g was weighed and subjected to extraction, MPLC fractionation, semi-HPLC purification and vacuum dehydration at 40 oC overnight, yields for the peak I and peak II were 4.2 and 7.6 mg and aliquots of the pure compounds were dissolved in d-methanol and subjected to 1H and 13C NMR analyses for structural elucidation. For peak I, the NMR spectra are shown in Supplementary Figure 1 with resulted data obtained as: 1H NMR (500 MHz, Methanol-d4) δ 7.83 (dd, J = 8.9, 3.0 Hz, 1H), 6.87 – 6.86 (m, 1H), 6.85 – 6.84 (m, 2H), 6.61 (d, J = 3.4 Hz, 1H), 4.09 – 4.06 (m, 1H), 3.89 – 3.87 (m, 2H), 3.63 (dd, J = 12.1, 6.5 Hz, 1H), 3.48 (d, J = 11.7 Hz, 1H), 3.45 (dd, J = 9.2, 3.2 Hz, 1H), 3.40 (t, J = 9.1 Hz, 1H), 3.31 (t, J = 9.4 Hz, 1H). 13C NMR (126 MHz, Methanol-d4) δ 184.14, 166.75, 164.19, 162.99, 161.49, 158.81, 129.68, 122.96, 117.07, 104.28, 102.67, 102.20, 95.25, 82.27, 80.29, 78.49, 77.77, 77.53, 75.11, 74.58, 72.80, 71.29, 63.35, 62.63, 62.13. In reference to the reported literatures [11,16], it was elucidated as saponarin (Figure 4A). In further, ESI-MS analysis (Figure 5A) showed a molecular ion at m/z 593.26. In the negative mode electrospray spectrometry (ESI-MS)/MS analysis (Figures 5A2), the fragment ion at m/z 431 gave the evidence for an O-linked hexose and the fragment ion ([MH-120]: m/z 473 suggested the presence of Y1 cleavage of a hexose (Figure 5A2). With the combination of fragmentations, the ion m/z 311 indicated the presence of elimination of one O-hexose and one C-hexose units. With reference to the reported dada [11], peak I was instrumentally elucidated and identified as saponarin of apigenin-6-C-β-D-glucopyranosyl-7-O-β-D-glucopyranoside and assigned as C27H30O15 (Figure 4A).
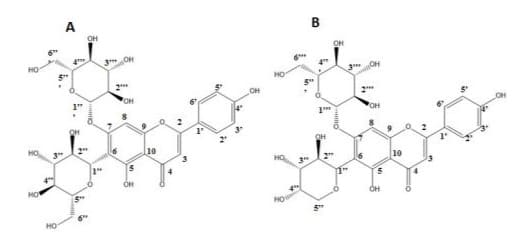
Figure 4: Chemical formula of A: orchid peak I: saponarin and B: peak II: apigenin-6-C- ribosyl-7-O-glucoside (ribosnarin).
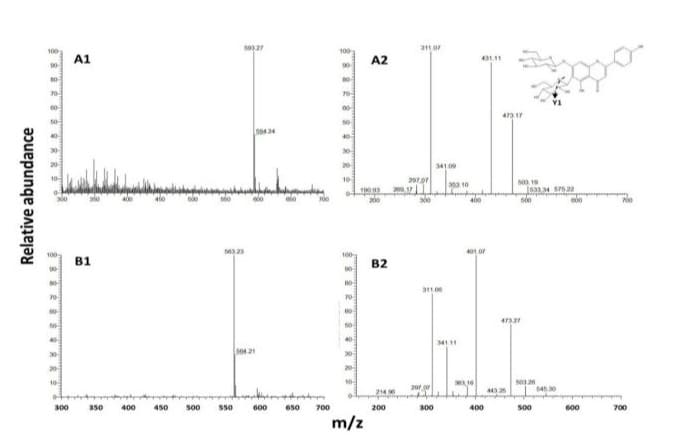
Figure 5: ESI-MS spectrometry analysis of orchid peak I (A) and peak II (B); 1: ESI-MS and 2: (ESI-MS)/MS and Y1 cleavage of a hexose of peak I shown at upper right corner.
For the peak II, the 1H and 13C NMR spectra are respectively shown in Figures 3A and 3B. The 13C-DEPT and COSY spectroscopic data are presented in Supplementary Figures 2 and 3. The resulted data obtained as: 1H NMR (500 MHz, Methanol-d4) δ 7.80 (dd, J = 8.9, 3.0 Hz, 1H), 6.97 - 6.94 (m, 1H), 6.86 - 6.80 (m, 2H), 6.59 (d, J = 3.4 Hz, 1H), 3.95 – 3.89 (m, 1H), 3.86 - 3.81 (m, 2H), 3.63 (dd, J = 12.1, 6.5 Hz, 1H), 3.58 (d, J = 11.7 Hz, 1H), 3.48 (dd, J = 9.2, 3.2 Hz, 1H), 3.42 (t, J = 9.1 Hz, 1H), 3.29 (t, J = 9.4 Hz, 1H). 13C NMR (126 MHz, Methanol-d4) δ 184.21, 166.75, 164.80, 162.99, 161.47, 158.82, 129.69, 122.95, 117.08, 111.78, 106.99, 104.25, 104.02, 96.35, 78.64, 76.96, 76.37, 75.17, 74.83, 71.73, 71.63, 71.03, 70.07, 62.73. In referenced to the reported data indicating that δ 62.7, 71.6, 74.8, 76.9, 78.6, and 104.0 are correlating to the glucosyl carbons and δ 70.1, 71.0, 71.7, 75.2, and 76.4 are correlating to the ribosyl carbons [6, 9,17], it is apparent that peak II bears glucosyl and ribosyl moieties. In further elucidation with ESI-MS analyses, a molecular ion at m/z 563.25 was detected (Figure 5B1). In the followed (ESI-MS)/MS analysis (Figure 5B2), the fragment ion at m/z 401 gave the evidence for an O-linked hexose and the fragment ion ([M-H-120]: m/z 473 suggested the presence of Y1 cleavage of a pentose. In further referenced to the reported literature [9], peak II was instrumentally elucidated as apigenin-6-C-β-D-ribopyranosyl-7-O-β-D-glucopyranoside, assigned as C26H28O14 and named “ribosnarin” (Figure 4B). In molecular comparison of the two identified compounds, the main structural difference between peak I and peak II is C-glucoside for the former and C-riboside for the latter. This featured difference deserves further properties characterization addressing on the structure-activity relationship in reflection of one -CH2O unit difference between saponarin and ribosnarin (Figure 4).
As done further, the purified samples were used as authentic references to construct standard curves and applied for estimation of their contents in the 11 collected white color moth orchids. The petals, sepals and lips were removed from each flower and pooled for drying and pulverized into powders for extraction and HPLC quantification. Abundant contents of peak I and peak II with ranges in 1.1 - 1.7% and 4.6 - 6.7% and averaged 1.5±0.5 and 5.6±1.1 % (dry basis) were determined. From each sample, the peak II content was higher than that of peak I. All white color moth orchid flowers were collected from the local flora nurseries with varied size and flower number per flower stem. The detected contents of saponarin and ribosnarin varied in a limited range as affected by different sources. This was in agreement with a previous report with red color moth orchid that about 0.58% (wet basis) of the peak II was detected [9]. For saponarin investigated as a viable chemotaxonomic marker [18,19], its synthesis as one of the secondary metabolites and localization in the mesophyll vacuole or leaf epidermis are largely stimulated by biotic and abiotic stresses. An appropriate biotic or abiotic approach could be a strategy to enhance saponarin biosynthesis. Saponarin is bioactive and has been potentially used to treat oxidative and inflammatory disorders, such as in protection against liver diseases, and reducing blood glucose, along with antiobesity effects [13]. Since peak II (ribosnarin) is a new emerging C-glycosylflavonid and its applicable potency has been less investigated, based on the intimate molecular structures, it could be regarded as a potential saponarin analog and both might function equally or synergistically one another. All these are of interest and deserve further investigations.
ABTS∙⁺ scavenging and Tyrosinase Inhibitory Activities Determination
For ABTS∙⁺ scavenging activity determination, they were concurrently run with authentic Trolox as a control (Figure 6) with concentrations ranged from 0 to 100 µM. The activities expressed by absorbance at 734 nm decreased with an increase of test concentration. At the concentrations of 50 and 100 µM, ABTS∙⁺ scavenging activities of Trolox was apparently higher than that of peak I or peak II. At each test concentration, ABTS∙⁺ scavenging activity of peak I and peak II varied in a very limited range. This indicates that ABTS∙⁺ scavenging activities as affected C-glucosyl and C-ribosyl moieties for the apigenin flavones is limited. Thus, peak II (ribosnarin) could be regarded as a novel candidate as saponarin belonging to the family of C-glycosylflavonoids to bear the same potencies of antioxidant and related biological activities [2,4,11,13,16].
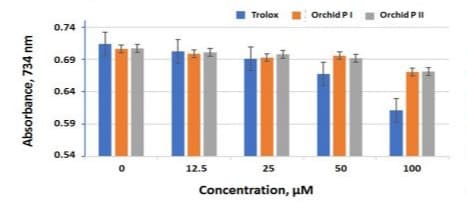
Figure 6: ABTS∙⁺ scavenging activities of orchid peak I and peak II and Trolox at various concentrations at 0, 12.5, 25, 50 and 100 µM dissolved in 20% DMSO; P I: orchid peak I and P II: orchid peak II.
For tyrosinase inhibitory activity determination, kojic acid as reference, orchid peak I and peak II were run concurrently by continuously monitoring absorbance increase at 490 nm (Figure 7). The inhibitory activity was evaluated based on the obtained slopes of the regression linear equations and coefficients (R2). All regression coefficients (R2) were higher than 0.99. For kojic acid (Figure 7A), a known tyrosinase inhibitor, slopes decreased remarkably from 0.0051 to 0.0016 with an increase of kojic acid concentrations from 0 to 100 µM. For the orchid peak I and Peak II (Figures 7B and C), the slopes deviated independently from
0.0065 to 0.0067 with the test concentrations. It is apparent that tyrosinase inhibitory activities for both peak I and peak II were considerably low in the test concentrations. This partially agreed with the observation of Lam et al. [9] who reported that peak I (saponarin) exhibits low while peak II (ribosnarin) bears potent tyrosinase inhibitory activity. For natural flavonoids, mushroom tyrosinase inhibitory activities is governed by 4-hydroxyl group and enhanced by additional hydroxyl group on 3- or 5-carbon position [20]. Thus, more obvious inhibitory activities might be detected at the higher concentrations then those tested in this study. And it is comprehensive to accept the observation that tyrosinase inhibitory activity as affected C-glucosyl and C-ribosyl moieties for the apigenin flavones is negligible.
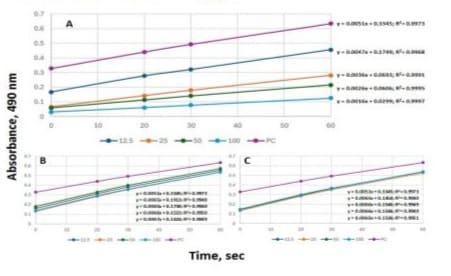
Figure 7: Tyrosinase activity determination by continuously measuring absorbance at 490 nm for 60 sec as affected by kojic acid (A), orchid peak I and peak II at 0, 12.5, 25, 50 and 100 µM and absorbance vs 0, 20, 30 and 60 sec of reaction were applied for construction of linear equations to show slopes and regression coefficients (R2).
HPLC Determination of β-Glucosidase Substrate Availability
Based on the structures of peak I and peak II both containing 7-β-D-glucoside moiety, the moiety might be hydrolyzed by almond β-glucosidase. In HPLC analysis of pNPG as a substrate for β-glucosidase hydrolysis, a specific peak monitored at 254 nm was detected before and after enzymatic hydrolysis (Figures 8A1 and A2). This indicates that HPLC analysis could be a novel means in determination of β-glucosidase activity with pNPG as a substrate. When orchid peak I and peak II were mixed and subjected to β-glucosidase hydrolysis, the original peaks were not changed (Figure 8B1). Interestingly, when either peak I or peak II was respectively mixed with pNPG and subjected to β-glucosidase hydrolysis, only pNPG was completely hydrolyzed after incubation at 60 oC for 10 min (Figures 8B2 and B3). It is obvious that both orchid peak I and peak II were not almond β-glucosidase hydrolysable. This was not in agreement with isosaponarin reported [21]. It is likely that isosaponarin bearing β-O-D-glucoside bond linking to 4’-carbon position which could be hydrolyzed by β-glucosidase. It is also apparent that either peak I or peak II did not exhibit hindrance or competitive inhibition against pNPG hydrolysis by the β-glucosidase. Based on the observation that activities of β-glucosidases from thermophilic fungus Myceliophthora heterothallica are affected by pH, temperature and chemicals have been observed [15], different sources of β-glucosidases may have varied catalytic characteristics. As noticed, even both saponarin and ribosnarin structures containing 7-β-D-glucoside bonds, which are not hydrolysable by almond β-glucosidase. The substrate availability was not altered by either linking to a 6-C-glucosyl or 6-C-ribosyl moiety. From the point of view of plant physiology, even the realistic nature for copresence of these two forms of C-glycosylflavoids have not been characterized, this observed properties remain research interest for further investigation.
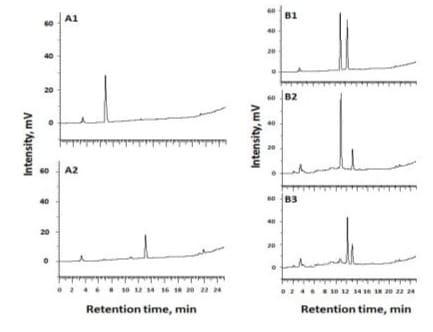
Figure 8: HPLC chromatograms of p-nitropheyl-β-Dglucopyranoside (pNPG) before and after β-glucosidase hydrolysis and β-glucosidase hydrolysis of mixtures of orchid peak I and peak II, peak I and pNPG and peak II and pNPG; A1: pNPG after reacted with heat inactivated β-glucosidase; A2: pNPG after reacted with β-glucosidase; B1: mixture of peak I and peak II after reaction; B2: mixture of peak I and pNPG after reaction; and B3: mixture of peak II with pNPG.
As concluded, two C-glycosylated flavonoids have been isolated and identified in the moth orchid flowers. Even O-glycosylated and C-glycosylated flavonoids are widespread in human diets while the latter are lack of interaction with metabolic hydrolysis and some are secreted in human urine following oral administration [1,4]. It is of interest to raise the wonder that hydrolysis of the 7-O-ß-D-glucosidic linkage was largely inhibited by the orthopresence of 6-C-glucoside or 6-C-riboside. Therefore, it is of merit to demonstrate that abundant quantities of the two C-glycosylated flavones up to 1.5 and 5.6% (dry basis) exist in moth orchid flowers which could be regarded as a potent source for further discovery of known and unknow purposes. Based on the assessed activities, saponarin and ribosnarin almost exhibited identically and differentiated negligibly by impact of 6-C-ribo- and 6-C-gluco- moiety. Ribosnarin, even has been less investigated, could be regarded as a natural analog of saponarin and integrated both as nutraceutical ingredients for value-added products development. Especially, nature for abundant presence of these two forms of C-glycosylflavoids in the moth orchid flowers deserve further investigations.
Supplementary Materials: The Supplementary Material is available free of charge including 1H and 13C NMR spectroscopic spectra of orchid peak I and 13C DEPT and COSY spectroscopic analyses of orchid peak II.
Acknowledgments: This research partially funded by the National Science Council, ROC (MOST 111-2320-B-415-003) is acknowledged. The publication is in memorial of Ms Bi-Yuan LiuHuang, Department of Horticulture, National Chiayi University, for her generous providing moth orchid cultivars and technical assistance.
Conflicts of Interest: The authors declare no conflict of interest.
References
- Rauter AP, Lopes RG, Martins A (2007) C-Glycosylflavonoids: identification, bioactivity and synthesis. Natural Product Communication 2: 1175-1196.
- Xie L, Deng Z, Zhang J, Dong H, Wang W, et al. (2022) Comparison of flavonoid O-glycoside, C-glycoside and their aglycones on antioxidant capacity and metabolism during in vitro digestion and in vivo. Foods 11: 882.
- Shie JJ, Chen CA, Lin CC, Ku AF, Cheng TJR, et al. (2010) Regioselective synthesis of di-C-glycosylflavones possessing antiinflammation activities. Organic and Biomolecular Chemistry 8: 44514462.
- Courts FT, Williamson G (2015) The occurrence, fate and biological activities of C-glycosyl flavonoids in the human diet. Critical Review of Food Science and Nutrition 55: 1352-1367.
- Ling LF, Subramaniam S (2007) Biochemical analyses of Phalaenopsis violacea orchid. Asian Journal of Biochemistry 2: 237-246.
- Kuo PC, Chen GF, Yang ML, Wu TS (2010) High-performance liquid chromatography profiling of pigments from Phalaenopsis hybrids and their contribution to antioxidant and antityrosinase activities. Acta Horticulture 878: 89-95.
- Minh TN, Tuyen PT, Khang DT, Va Quan VN, Thu Ha PT, et al. (2017) Potential use of plant waste from the moth orchid (Phalaenopsis Sogo Yukidian “V3”) as an antioxidant source. Foods 6: 85.
- Minh TN, Khang DT, Tuyen PT, Minh LT, Anh LH, et al. (2016) Phenolic compounds and antioxidant activity of Phalaenopsis orchid hybrids. Antioxidants 5: 31.
- Lam SH, Huang HY, Yang ML, Chen HH, Kuo PC, et al. (2019) Chemical constituents from Phalaenopsis hybrids and their bioactivities. Natural Product Communications 14: 1-5.
- Nagai M, Akita K, Yamada K, Okunishi I (2010) The effect of isosaponarin isolated from wasabi leaf on collagen synthesis in human fibroblasts and its underlying mechanism. Journal of Natural Medicine 64: 305-314.
- Seo KH, Park MJ, Cha JH, Ra JE, Han SI, et al. (2014) Saponarin from barley sprouts inhibits NF-αB and MAPK on LPS-induced RAW 264.7 cells. Food and Function 5: 3005.
- Lu CW, Yeh KC, Chiu KM, Lee MY, Lin TY, et al. (2022) The effect of isosaponarin derived from wasabi leaves on glutamate release in rat synaptosomes and its underlying mechanism. International Journal of Molecular Science 23: 8752.
- Kantharaj A, Yoon YE, Lee KA, Choe H, Chohra H, et al. (2023) Saponarin, a di-glycosyl flavone from barley (Hordeum vulgare L.): An effective compound for plant defense and therapeutic application. ACS Omega 8: 22285-22295.
- Lin SM, Lin BH, Hsieh WM, Ko WC, Liu CD, et al. (2010) Structural identification and bioactivities of red-violet pigments present in Basella alba fruits. Journal of Agricultural and Food Chemistry 58: 1036410372.
- Silva VCT, Coto ALS, Souza RC, Neves MBS, Gomes E, et al. (2016) Effect of pH, temperature, and chemicals on the endoglucanases and
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.