miR-223-3p Regulates Endothelial Cell Senescence Through HDAC2 Targeting Mediated by Transcription Factor SOX9
by Hyo-Jin Kim1,2, Phan Thanh Nam4, Jeong-Hyung Lee4, Cheol Hwangbo1,3*
1Division of Life Science, College of Natural Sciences, Gyeongsang National University, Jinju 52828, Republic of Korea
2Plant Molecular Biology and Biotechnology Research Center (PMBBRC) and Research Institute of Life Sciences, Gyeongsang National University, Jinju 52828, Republic of Korea
3Division of Applied Life Science (BK21 Four), Research Institute of Life Sciences, Gyeongsang National University, Jinju 52828, Republic of Korea
4Department of Biochemistry (BK21 Four), College of Natural Sciences, Kangwon National University, Chuncheon, Gangwon 24414, Republic of Korea
*Corresponding author: Cheol Hwangbo, Division of Life Science, College of Natural Sciences, Gyeongsang National University, Jinju 52828, Republic of Korea.
Received Date: 20 May, 2025
Accepted Date: 26 May, 2025
Published Date: 28 May, 2025
Citation: Kim HJ, Hwangbo C, Nam PT, Lee JH (2025) miR-223-3p Regulates Endothelial Cell Senescence Through HDAC2 Targeting Mediated by Transcription Factor SOX9. Int J Geriatr Gerontol 9:203. https://doi.org/10.29011/2577-0748.100203
Abstract
Toll-like receptor 4 (TLR4), that the innate immune response receptor, critically regulates endothelial cell senescence through the modification of histone acetylation by HDAC2 in age-induced emphysema. However, the molecular mechanisms by which HDAC2 regulates aging and cellular senescence remain unclear. Using cigarette smoke extract (CSE), the primary cause of emphysema, we demonstrate that TLR4 and HDAC2 expressions are reduced, and cellular senescence is induced in response to CSE in human umbilical vein endothelial cells (HUVECs), akin to emphysematous endothelial cells. The expression levels of miR223 in chronic obstructive pulmonary disease (COPD) patients and normal lung tissues were analyzed using bioinformatics. We identified miR-223-3p as the key regulating factor of cellular senescence through direct targeting of HDAC2 in HUVECs. MiR223-3p mimics increase p16INK4A expression, subsequently leading to cellular senescence, whereas silencing miR-223-3p decreases HDAC2 and p16INK4A expression, thereby preventing cellular senescence in the context of endothelial cell aging. Activated SOX9 in CSE-treated or TLR4 siRNA-treated cells regulates cellular senescence by modulating miR-223-3p expression. These findings demonstrate that SOX9 and miR-223-3p act as important regulators of cellular senescence by targeting HDAC2, thus providing novel insights into the previously unrecognized TLR4-HDAC2 cellular senescence mechanism in endothelial cells.
Key words: miR-223-3p, SOX9, cell senescence, emphysema, HDAC2
Introduction
Cellular senescence is defined as the cessation of cell cycle and is caused by various intrinsic and extrinsic stresses such as telomere shortening, oncogenic activation, radiation or oxidative damage [1-3]. The cell cycle arrest by senescence is regulated by the cyclin-dependent kinase inhibitors, including p53/p21WAF1/CIP1 and p16INK4A/pRB, and overexpression of any of these may induce cell senescence [4, 5]. Senescent cells secrete senescence-associated secretory phenotype (SASP), including cytokine, chemokine and matrix metalloproteinases, which can be transmitted to surrounding cells and induce aging of normal cells [6, 7]. Senescence associated beta-galactosidase (SA-β-gal) that a lysosomal hydrolase that is activated by enhanced lysosomal biosynthesis, and expanding morphology resulting from accumulation of cytoplasmic granules in the lysosomes are a representative phenotype of senescent cells [8, 9]. Cellular senescence contributes significantly to organ dysfunction, tissue impairment, and various age-related diseases [10].
Toll-like receptor 4 (TLR4) is a representative pattern recognition receptor that recognizes pattern molecules, such as Lipopolysaccharid, and induces innate immune responses [11]. Recent studies suggest that changes in TLR4 expression correlate with age-related diseases [12]. In particular, a deficiency of TLR4 in hyperoxia increases lung injury and mortality despite the innate immune function of TLR4 [13, 14]. Notably, TLR4 knockout mice develop emphysema after 3 months of age even without other external stimuli [15]. It has been established that emphysema caused by TLR4 down-regulation leads to increased expression of p16INK4a through histone deacetylase 2 (HDAC2) reduction and appears to be dependent on endothelial cell senescence [16]. However, the molecular regulatory mechanisms of HDAC2 involved in endothelial cell senescence remain unknown.
Sry-related HMG-box 9 (SOX9), a transcription factor crucial for male gender determination, belongs to the SOX family of genes that contain a high mobility group box [17]. SOX9 influences lung branch morphogenesis in lung epithelial cells, and its deletion leads to issues like disrupted cellular architecture and abnormal cell migration [18]. Microarray data analysis showed increased SOX9 expression in elderly skin tissues compared to young people [19]. Moreover, increased expression of SOX9 in vascular smooth muscle cells induces features of cell senescence, such as decreased cell proliferation, increased DNA damage [20]. However, the mechanism by which SOX9 regulates cellular senescence remains unclear.
The expression of various miRNAs vary with age, suggesting the possibility that miRNAs may be involved in senescent pathophysiology [21]. These changes in miRNAs are also observed in the lungs of mice following aging or cigarette smoke exposure [22]. Our study proposes that miR-223-3p regulates HDAC2 expression in the TLR4-mediated senescence mechanism. We found miR-223-3p using bioinformatics analysis of GSE database. MiR-223-3p directly binds to the HDAC2 3’-UTR, inhibiting its expression in human umbilical vein endothelial cells (HUVECs). The reduction of HDAC2 expression by miR-223-3p increases p16INK4a expression, subsequently affecting cell cycle arrest and cell senescence. SOX9 binds to the miR-223-3p promoter, enhancing its expression and thereby regulating endothelial cell aging. These results provide the foundation for the novel role of SOX9 and miR-223-3p in TLR4-mediated senescence mechanism in endothelial cells.
Results TLR4 reduction or CSE induces cellular senescence in human endothelial cells
The reduction of TLR4 expression leads to cellular senescence in endothelial cells [16]. Inhibition of TLR4 in HUVECs leads to increased expression of p16INK4a, a protein involved in aging regulation (Figure 1A). SA-β-gal staining, a common marker for senescent cells, revealed increased activity in siTLR4-treated cells (Figure 1B). Subsequently, we confirmed changes in the SASP following TLR4 knockdown. This resulted in increased expression of IL-1a, IL-6, IL-8, and GM-CSF (Figure 1C).
The cigarette smoke causes DNA damage, airway injury, and increased cell senescence in the lungs [23]. We previously reported that TLR4-HDAC2 signaling is important for lung endothelial cell senescence [16]. To investigate the correlation between smoking and the expression of TLR4 and HDAC2, we utilized cigarette smoke extract (CSE) obtained in our laboratory. With longer processing time of CSE, the protein expression of TLR4 and HDAC2 decreased (Figure 1D). Additionally, the protein expression declined with increasing treatment concentration of CSE (Figure 1E). Our previous data demonstrated that p16INK4a expression is regulated by TLR4 expression. To confirm, we measured the expression of p16INK4a during CSE treatment at specified time points and concentrations and found that enhanced p16INK4a expression compared to control (Figure 1F). Notably, p16INK4a expression significantly decreases when the CSE treatment time exceeds 8 h (Figure S1A). The reduced p16INK4a expression seems to correlate with cell death during prolonged CSE treatment (Figure S1B). Cellular senescence was more pronounced in CSEtreated cells compared to control cells (Figure 1G). G1 phase arrest is a characteristic of downregulation of TLR4 induced cell cycle arrest in previous study. TLR4 knockdown cells in G0/G1 phase increased (63.70% vs. 58.34%) while S (9.30% vs. 11.45%) and G2/M phase (25.16% vs. 27.68%) decreased compared to siCon (Figure S1C). Flow cytometry analysis for cell cycle distribution demonstrated that increased in G0/G1 phase (65.46% vs. 57.89%) in HUVECs with 5% CSE compared to untreated control (Figure S1D). These data suggested that CSE induced cell senescence through expression of TLR4, HDAC2 and p16INK4a in HUVECs.
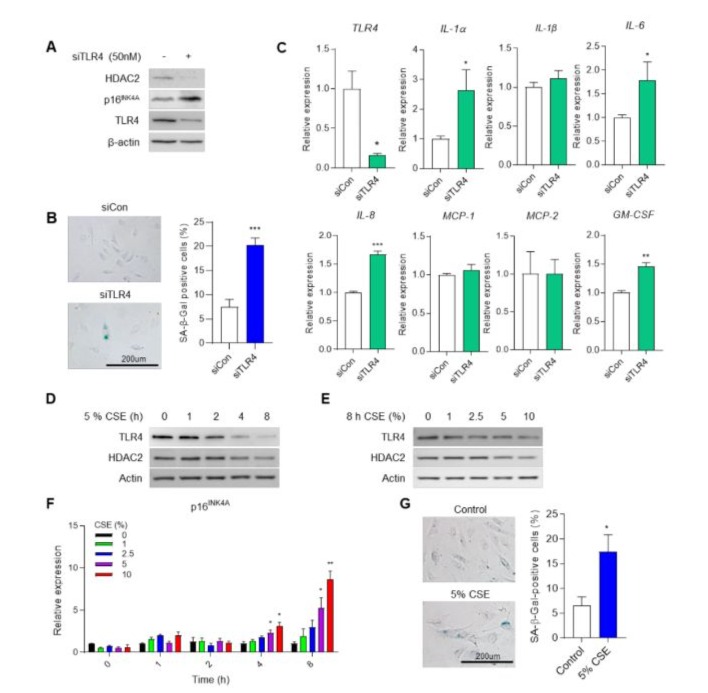
Figure 1: Knockdown of TLR4 and CSE induces cell senescence via p16INK4a expression. (A) HDAC2 and p16INK4a protein expression in control (siCon) or TLR4-knockdown (siTLR4) by siRNA in HUVECs. (B) SA-β-gal staining in siCon and siTLR4 cells. Quantification of the SA-β-gal staining. ***p <0.001 vs. siCon. (C) mRNA level of SASP using qRT-PCR. *p <0.05, ***p < 0.001 vs. siCon. TLR4 and HDAC2 expression in response to CSE in a (D) time-dependent and (E) dose-dependent manner in HUVECs. (F) Expression of p16INK4a mRNA in HUVECs at indicated time- and dose-dependent treatment of CSE. *p <0.05, **p < 0.01 vs. 0% at same treatment time (G) SA-β-gal staining Control or 5% CSE for 8 h treated cells. Quantification of the SA-β-gal staining. *p <0.05 vs. Control.
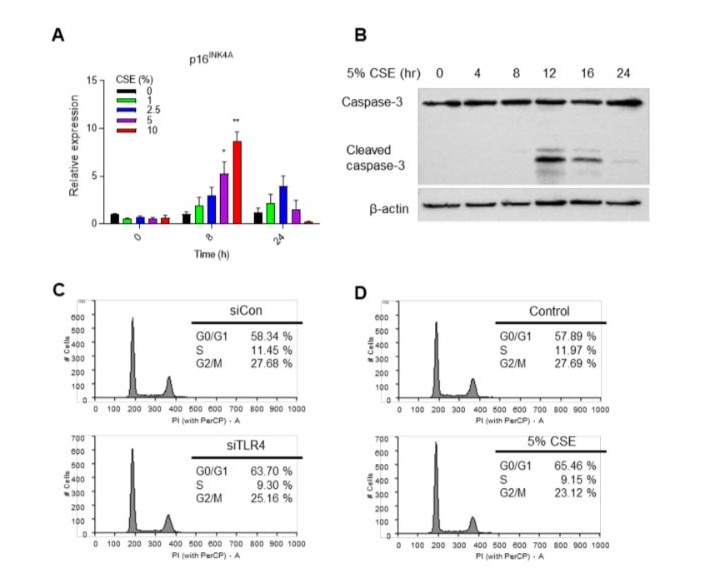
Supplementary Figure 1: Appropriate CSE treatment induces cell senescence not cell death. (A)Expression level of p16INK4a by CSE treated with the concentration and time indicated using qRT-PCR. (B) Western blot analysis of caspase-3 activation measured according to 5% CSE treated at the indicated times. (C) Flow cytometry analysis for cell cycle distribution of siCon or siTLR4 transfected HUVECs. (D) Flow cytometry analysis for cell cycle distribution of un-treated or 5% CSE treated HUVECs.
MiR-223 directly targets HDAC2 in endothelial cells
Although HDAC2 knockdown-mediated endothelial cell senescence is an important cause of chronic obstructive pulmonary disease (COPD), the regulator of HDAC2 is not yet known [16]. We investigated the expression levels of miRNAs and mRNAs in COPD patients compared to normal individuals using GSE 38974 data (Figure 2A). Our focus was on relatively changed miRNA expression, and we observed enhanced expression of certain miRNAs in COPD patients (Figure 2B). Notably, miR-223 exhibited the highest increase. MiR-223 has been identified as a regulator of granulocyte differentiation and has a reverse effect on erythroid differentiation [24]. To determine the miR-223-3p binding site, we performed in silico analysis and identified the HDAC2 3’-UTR (Figure 2C). Next, we evaluated HDAC2 mRNA levels in miR-223-3p-overexpressing HUVECs and control cells (Figure 2D). HDAC2 mRNA increased significantly in miR-223-3p-overexpressing HUVECs compared to control cells. In a previous study, we demonstrated that HDAC2 expression is crucial in emphysema, a typical symptom in COPD patients. Consequently, we found a clinically relevant correlation between HDAC2 and miR-223 expression in COPD patients (Figure 2E). However, in normal individuals, no significant correlation was observed between HDAC2 and miR-223 (Figure 2F). These results provide evidence that miR-223-3p directly regulates HDAC2 expression in endothelial cells.
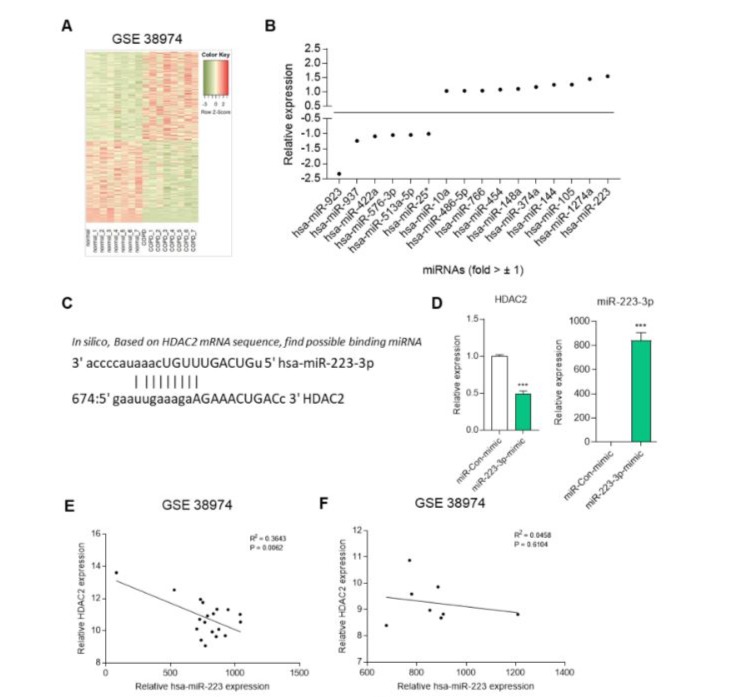
Figure 2: miR-223 directly targets HDAC2. Heat map analysis of gene expression between in normal and COPD patients. (B) The relative fold changes in miRNA in COPD patients compared to normal. (C) The predicted binding sites of miR-223-3p within the HDAC2 are presented. (D) Expression of HDAC2 mRNA in control (miR-con-mimic) or miR-223-3p-mimic transfected HUVECs. ***p < 0.001 vs. miR-con-mimic. (E) Correlation between HDAC2 and miR-223-3p expression in COPD patients using GSE 38974 dataset analysis. (F) Correlation between HDAC2 and miR-223-3p expression in normal using GSE 38974 dataset analysis.
MiR-223-3p-mimic induces endothelial cell senescence
To investigate the impact of TLR4 on miR-223-3p expression, we knocked down TLR4 using siRNA in HUVECs, resulting in an upregulation of miR-223-3p (Figure 3A). Additionally, miR-223 expression responded to 5% CSE exposure (Figure 3B). Next, we examined the expression of p16INK4a, a downstream signaling and senescence regulatory gene of TLR4. Overexpression of miR-2233p led to enhanced p16INK4a expression in HUVECs (Figure 3C). We also assessed SA-β-gal activity, a marker of cellular senescence, and observed an increase in positive blue-stained cells in miR-223-3p-overexpressing cells compared to control cells (Figure 3D). Furthermore, the growth of miR-223-3p-overexpressing cells declined relative to miR-con-mimic cells (Figure 3E). Cell cycle analysis revealed that miR-223-3p-transfected cells accumulated in the G0/G1 phase (63.15% vs. 55.13%), with decreased proportions in the S phase (10.73% vs. 11.36%) and G2/M phase (26.01% vs. 33.49%) compared to miR-con-mimic-expressing cells (Figure 3F). Collectively, these findings demonstrate that miR-223-3p plays a role in regulating p16INK4a expression and cellular senescence in HUVECs.
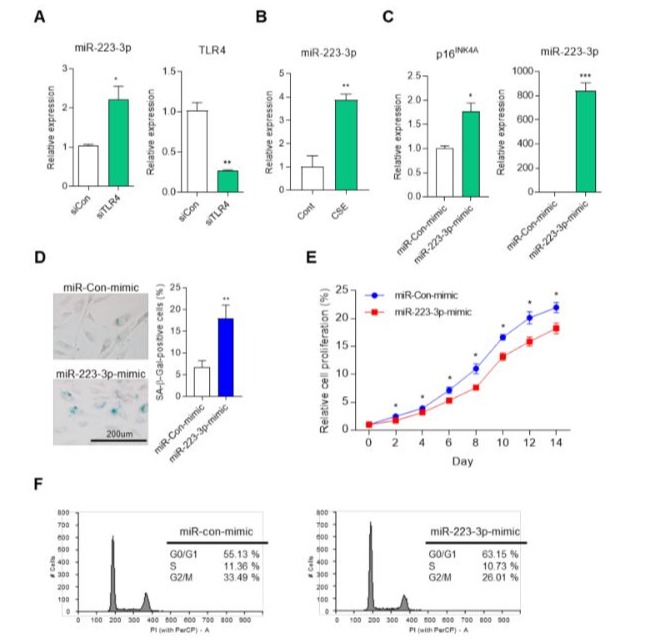
Figure 3: miR-223-3p-mimic regulates cell senescence in endothelial cells. (A) The expression of miR-223-3p in siCon or siTLR4 transfected HUVECs. *p <0.05, **p < 0.01 vs. siCon. (B) The expression of miR-223-3p in HUVECs after treated 5% CSE for 8hr. **p < 0.01 vs. Con. (C) The mRNA expression of p16INK4a in control (miR-Con-mimic) or miR-223-3p-mimic transfected HUVECs. *p <0.05, ***p < 0.001 vs. miR-Con-mimic. (D) SA-β-gal staining in miR-Con-mimic or miR-223-3p-mimic transfected HUVECs. Quantification of the SA-β-gal staining. **p < 0.01 vs. miR-Con-mimic. (E) The growth curve of miR-Con-mimic and miR-223-3pmimic transfected HUVECs were measured over 14 days. *p <0.05 vs. miR-Con-mimic. (F) Flow cytometry analysis for cell cycle distribution of miR-Con-mimic and miR-223-3p-mimic transfected HUVECs.
Anti-miR-223-3p prevents endothelial cell senescence
To support our hypothesis, we investigated alterations in HDAC2 and p16INK4a expression using antisense-miR-223-3p (anti-miR-2233p). In HUVECs overexpressing anti-miR-223-3p, HDAC2 expression increased, while p16INK4a expression decreased compared to anti-miR-con overexpressing cells (Figure 4A). Next, we examined cell proliferation in miR-223-3p-inhibited HUVECs under two senescence-inducing conditions: TLR4 downregulation and CSE treatment. Senescence-induced cells exhibited reduced proliferation compared to control cells, but anti-miR-223-3p overexpressing cells showed increased proliferation (Figure 4B and C). We also assessed p16INK4a protein expression and H4K8 acetylation (H4K8ac) levels under the same conditions. In TLR4 knockdown or CSE treatment conditions, p16INK4a expression and H4K8ac levels were elevated. However, when miR-223-3p was inhibited in senescence-inducing cells, p16INK4a expression and H4K8ac levels decreased relative to senescent cells (Figure 4D and F). Furthermore, anti-miR-223-3p repressed cellular senescence induced by TLR4 knockdown or CSE treatment, as evidenced by reduced SA-β-gal activity (Figure 4E and G). Flow cytometry analysis revealed that TLR4 knockdown cells exhibited an enhanced G0/G1 phase (64.26% vs. 58.39%) compared to siCon, which was prevented in anti-miR-223-3p-transfected cells (59.89% vs. 64.26%) (Figure 4H). Similarly, 5% CSEtreated cells showed increased G0/G1 phase (65.21% vs. 56.27%) compared to untreated controls, but anti-miR-223-3p overexpressing cells mitigated this increase (60.36% vs. 65.21%) (Figure 4I). Collectively, these data suggest that miR-223-3p plays a crucial role in the TLR4-HDAC2-p16INK4a signaling pathway in endothelial cells.
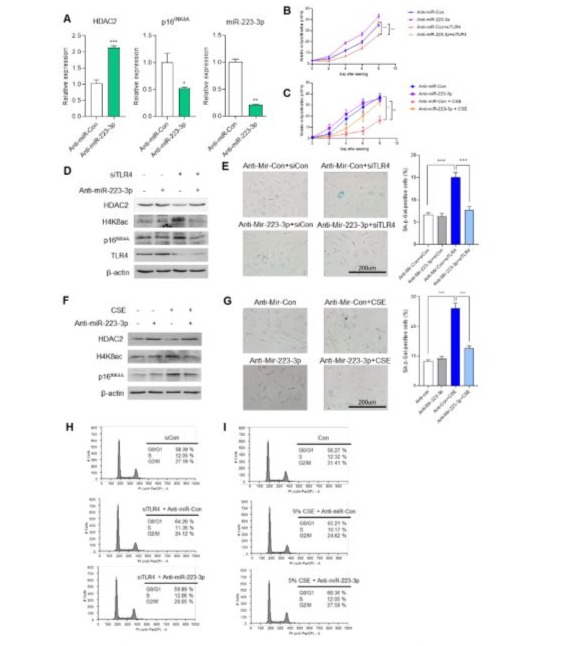
Figure 4: Anti-miR-223-3p prevents cell senescence induced by TLR4-knockdown or CSE. (A)The mRNA expression of HDAC2 and p16INK4a in control (anti-miR-Con) or anti-miR-223-3p transfected HUVECs. *p <0.05, **p < 0.01, ***p < 0.001 vs. anti-miR-Con. (B) The growth curve of siTLR4 and siCon HUVECs were transfected with anti-miR-Con or anti-miR-223-3p. After seeding 1x10^4 cells per well, cell growth was measured by counting every 2 days for 8 days. (C) The growth curve of siTLR4 and siCon HUVECs were treated with 5% CES for 8hr. After seeding 1x10^4 cells per well, cell growth was measured by counting every 2 days for 8 days. (D) Western blot analysis for measured HDAC2, H4K8ac and p16INK4a expression in indicated siRNA or anti-miR transfected HUVECs. (E) SA-β-gal staining in indicated siRNA or anti-miR transfected HUVECs. Quantification of the SA-β-gal staining. ***p < 0.001. (F) Western blot analysis for measured HDAC2, H4K8ac and p16INK4a expression in siTLR4 or siCon HUVECs with treatment of 5% CSE for 8hr. (G) SA-β-gal staining in siTLR4 or siCon HUVECs with treatment of 5% CSE for 8hr. Quantification of the SA-β-gal staining. ***p < 0.001. (H) Flow cytometry analysis for cell cycle distribution of indicated siRNA or anti-miR transfected HUVECs. (I) Flow cytometry analysis for cell cycle distribution of siTLR4 or siCon HUVECs with treatment of 5% CSE for 8hr.
SOX9 regulates endothelial cell senescence via miR-223-3p expression
To identify the expression regulator of miR-223-3p, we explored transcription factors using a prediction website (Figure S2A). Analysis in two datasets allowed us to anticipate the interaction of the promoter of miR-223-3p and transcription factor, SOX9. When we decreased SOX9 expression using siRNA in HUVECs, miR-223-3p expression was downregulated (Figure 5A). Additionally, we assessed promoter activity through a luciferase assay and observed increased activity in CSE-treated HEK 293T cells (Figure S2B). The decrease in TLR4 expression coincided with elevated SOX9 expression and phosphorylation of Ser181 in SOX9 (p-SOX9), the active form (Figure 5B). Furthermore, CSE-treated HUVECs showed induction of both SOX9 and p-SOX9 (Figure 5C). We investigated SOX9 localization using a fractionation assay, revealing a shift from the cytosol to the nucleus upon TLR4 reduction or CSE treatment (Figure 5D and F). Fluorescence staining confirmed SOX9 translocation to the nucleus under these conditions (Figure 5E and G). Next, we examined downstream signaling and cellular senescence changes resulting from decreased SOX9 expression under aging-induced conditions. CSE treatment led to decreased HDAC2 expression and increased H4K8ac and p16INK4a levels, which were counteracted by SOX9 reduction via siRNA (Figure 5H). Moreover, SOX9 reduction attenuated the upregulation of miR-223-3p induced by CSE treatment (Figure 5I). Cellular senescence induced by CSE was mitigated by SOX9 reduction, as observed in SA-β-gal staining experiments (Figure 5J). Additionally, treating HUVECs with JQ1, a specific SOX9 inhibitor, above 1 μM resulted in decreased SOX9 protein expression and increased p16INK4a levels (Figure S2C). JQ1 treatment also counteracted the reduction of HDAC2 and the elevation of H4K8ac and p16INK4a caused by CSE (Figure S2D). Furthermore, JQ1 treatment reduced miR-223-3p expression induced by CSE (Figure S2E) and diminished CSE-induced cellular senescence (Figure S2F). In summary, our findings demonstrate that SOX9 plays a crucial role in regulating endothelial cell senescence through miR-223-3p expression.
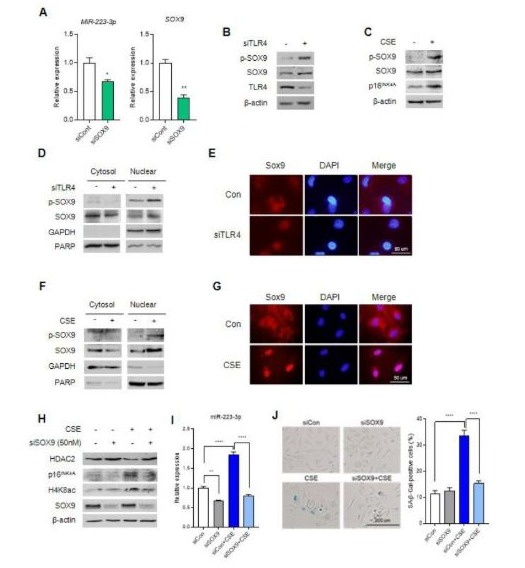
Figure 5: SOX9 promotes endothelial cell senescence via miR-223-3p. (A)The expression of miR-223-3p in siCon or siSOX9 transfected HUVECs quantified using qRT-PCR. *p <0.05, **p < 0.01 vs siCon. (B) Western blot analysis for measured p-SOX9, SOX9 and TLR4 in siCon or siTLR4 transfected HUVECs. (C) Western blot analysis for measured p-SOX9 and SOX9 in HUVECs with 5% CSE treatment for 8 h. (D) Cytosol and nuclear fractionation analysis for measured SOX9 and p-SOX9 localization in siCon or siTLR4 transfected HUVECs. (E) SOX9 localization in siCon or siTLR4 transfected HUVECs using fluorescence experiment. (F) Cytosol and nuclear fractionation analysis for measured SOX9 and p-SOX9 localization in HUVECs with 5% CSE treatment for 8 h. (G) SOX9 localization in HUVECs with 5% CSE treatment for 8 h using fluorescence experiment. (H) Western blot analysis for measured HDAC2, H4K8ac and p16INK4a expression in indicated CSE or siSOX9 treated HUVECs. (I) The expression of miR-223-3p level in indicated CSE or siSOX9 treated HUVECs using qRT-PCR. (J) SA-β-gal staining in indicated CSE or siSOX9 treated HUVECs. Quantification of the SA-β-gal staining. ***p < 0.001.
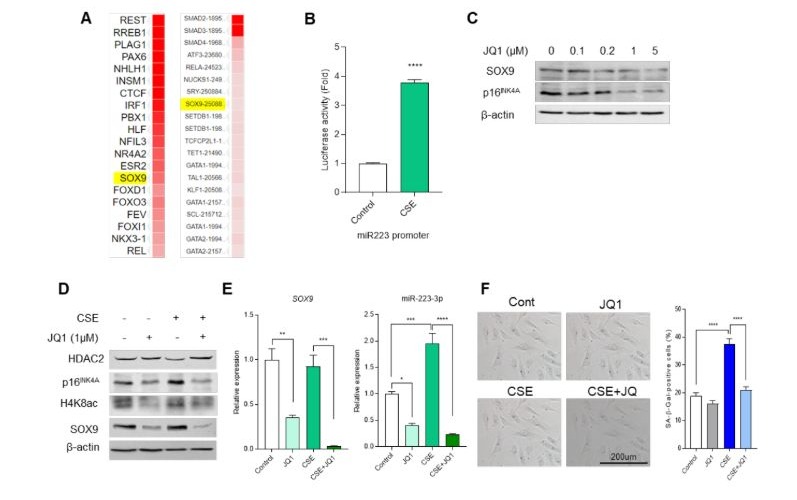
Supplementary Figure 2: SOX9 inhibitor, JQ1, regulates cell senescence. (A)List of transcription factors that can bind to the promoter of miR-223-3p found by in silico analysis (B) Analysis of luciferase promoter activity confirming the change in promoter activity of miR-223-3p after treatment with 5% CSE for 8 h in HEK293T cells. Quantification of luciferase activity. ***p < 0.001 vs. Con. (C) Western blot analysis confirming the decrease in expression of SOX9 when JQ1 was treated for 24 h by concentration. (D) Western blot analysis for measured HDAC2, H4K8ac and p16INK4a expression in indicated CSE or JQ1 treated HUVECs. (E) mRNA expression levels of SOX9 and miR-223-3p following CSE and JQ1 treatment using qRT-PCR. Quantification of mRNA expression. ***p < 0.001. (F) SA-β-gal staining in indicated CSE or JQ1 treated HUVECs. Quantification of the SA-β-gal staining. ***p < 0.001.
Discussion
Cellular senescence, characterized by irreversible cell cycle arrest, has traditionally been associated with cancer suppression. However, recent research suggests that it also disrupts tissue integrity, impairs organ function, and contributes to age-related diseases [10]. As our bodies age, most organs experience functional decline [25]. The aging lung undergoes structural changes, loss of function, and increased inflammation, accelerated lung senescence is a key pathological feature of COPD [26]. CSE is a well-known activator of cellular senescence, leading to decreased cell growth and altered expression of senescence marker proteins such as pRB and p16INK4a [27]. Notably, p16INK4a expression is closely linked to aging-related diseases and contributes to emphysema phenotypes, including airspace enlargement in both human epithelial cells and mice [28, 29]. CSE-induced premature lung aging results from increased oxidative stress, inflammatory responses, DNA damage, and the accumulation of senescent cells [27]. Chronic smokers experience rapid lung function decline, largely driven by cellular senescence [30].
Related to the molecular mechanism of senescence phenomenon, thus we proposed that the novel signaling of emphysema development via TLR4 [16]. Additionally, HDAC2 expression is reduced in TLR4-deficient mice, a finding consistent with observations in smokers and COPD patients [31], and HDAC2 regulates p16INK4a expression through H4K8 acetylation in the p16INK4a gene. Furthermore, we investigated the role of SOX9 and miR-223-3p in endothelial cell senescence, specifically related to TLR4 inhibition. Our findings suggest that these aging regulators also play a crucial role in CSE-induced endothelial cell senescence.
Histone acetylation plays a crucial role in gene expression regulation. By reducing the positive charge of histones through acetylation, their interaction with the negatively charged DNA phosphate groups is weakened. Consequently, chromatin structure is relaxed, allowing for increased gene transcription [32]. HDAC2, a member of Class I HDACs, removes acetyl groups from lysine residues in core histones (such as H2A, H2B, H3, and H4) [33]. In severe COPD patients and smokers, HDAC expression and activity are decreased compared to normal and non-smokers. Notably, HDAC2 expression is significantly altered [31]. When HDAC2 expression is downregulated using inhibitors or siRNA, alveolar septal cell loss occurs—a hallmark phenotype of emphysema—in human pulmonary microvascular endothelial cells and mice [34]. Interestingly, HDAC2 expression accumulates in senescent cells compared to young cells. This change is associated with altered p16INK4a expression and cell proliferation [35]. HDAC2 knockout mice exhibit an increased cellular senescence response to cigarette smoke, while HDAC2 overexpression does not have the same effect. HDAC2 also influences cell cycle progression by regulating the expression of cell cycle-related proteins and facilitating the transition from G1 to S phase [36, 37]. Despite these insights, the precise regulatory factors governing HDAC2 expression remain unclear.
To shed light on HDAC2 target regulatory factor, we found out miR223 using bioinformatics analysis. Our findings highlight miR223-3p as the most significantly accumulated factor in this context. MiR-223 was initially identified as a regulator of granulocyte differentiation in the hematopoietic system [24]. Its roles in human diseases include repression of stmn1 in hepatocellular carcinoma and inhibition of E2F1 in acute myeloid leukemia [38]. Increased miR-223 expression has been associated with age-related diseases such as Parkinson’s disease and diabetes [39]. Notably, miR-223 in pulmonary cells regulates HDAC2 expression and chemokine levels [40]. However, the molecular mechanism of miR-223 in cellular senescence remained elusive. In our study, we confirmed that miR-223-3p plays a central role in endothelial cell senescence. Overexpression of miR-223-3p in HUVECs increased p16INK4a expression and induced cell senescence. Conversely, reducing miR-223-3p using anti-miR-223-3p led to increased HDAC2 expression and decreased p16INK4a expression, suggesting that miR-223-3p modulates HDAC2 levels and contributes to cell senescence via TLR4-mediated signaling.
Transcription factors play a crucial role in gene expression regulation and are intricately linked to the aging process [41]. Among these factors, SOX proteins, which contain the HMG box from the male sex determinant gene sry, exhibit varying expression patterns during aging. Interestingly, inhibiting the expression of SOX2 and SOX4 leads to pathological symptoms associated with premature aging [42, 43]. SOX5, another member of the SOX family, induces p19Arf-dependent acute cell senescence in glioma cells [44]. In colorectal cancer, SOX9 promotes cell growth, suppresses cellular senescence, and enhances tumor growth and metastasis [45]. Conversely, overexpressing SOX9 in vascular smooth muscle cells results in senescence-like features, including impaired cell proliferation and increased DNA damage [20]. Notably, SOX9 exhibits tissue-specific expression patterns during aging [46]. Regarding apoptosis, elevated SOX9 exacerbates myocardial cell apoptosis under hypoxic conditions by modulating miR-223-3p and inhibiting MEF2C transcription [47]. Additionally, SOX9 overexpression in human bronchial epithelial cells exposed to CSE inhibits cell death [48]. Our findings reveal that SOX9 acts as a novel regulator of HDAC2/p16INK4a-mediated endothelial cell senescence by targeting miR-223-3p. Inhibition of SOX9 expression using siRNA and inhibitors delays cellular senescence in a TLR4-dependent and CSE-treated context.
In our study, we propose that HDAC2 expression is regulated by SOX9 and miR-223-3p within a TLR4-mediated senescence mechanism. Specifically, miR-223-3p directly binds to the 3’UTR of HDAC2, leading to its inhibition in HUVECs. This reduction in HDAC2 expression subsequently increases p16INK4a levels, contributing to cell cycle arrest and cellular senescence. Additionally, SOX9 binds to the promoter region of miR-223-3p, enhancing its expression and thereby influencing endothelial cell aging. Our findings shed light on the intricate relationship between cell senescence and age-related diseases. Furthermore, targeting SOX9 or miR-223-3p may hold therapeutic promise in managing these conditions.
Materials and Methods Cell culture and reagents
HUVECs were purchased from Lonza (C2519A, Basel, Switzerland) and maintained in M199 (sigma, M4530) supplemented with penicillin-streptomycin (17-602E, Lonza), 30 μg/ml ECGS (354006, Discovery labware, MA, USA), 100 μg/ ml Heparin (H3149, Sigma, Massachusetts, USA) and 20% fetal bovine serum (1040, ATCC, Washington, USA). HEK 293T cells were purchased from Korean cell line bank (21573, Daejeon, Republic of Korea) and cultured in Dulbecco’s Modified Eagle’s Medium (LM 001-05, Welgene, Gyeongsan-si, Republic of Korea) supplemented with 10% fetal bovine serum and 1% PenicillinStreptomycin mixture (17-602E, Lonza). Cells were maintained in a humidified 5% CO2 atmosphere at 37 °C.
CSE was used in this research. Briefly, connect one side of the tube with a research cigarette (3R4F Composition, University of Kentucky) and immerse one side in a 10ml M199 media. Install a pump to induce smoke to move inside when burning a cigarette.
While the cigarette combusted, a pump removed air from the tube, drawing cigarette smoke into the media and filtered with 32mm syringe filter with 0.45um supor membrane (4654, Pall, New york, United States). The 10ml solution made when all of one cigarette was burned was measured with 100% CSE.
RNA interference experiments
Negative control siRNA was purchased from Bioneer (O-2402210005, Daejeon, Republic of Korea). TLR4 or SOX9 targeting siRNA were purchased from Thermo Fisher (HSS110819, HSS110100, Massachusetts, USA). Transfection of siRNA oligo duplex was accomplished using Lipofectamine RNAiMAX
(13778-150, Invitrogen, California, USA) followed by instructions of manufacturers. After incubation for 48 h in HUVECs, the cells were gained, and knockdown efficiency was confirmed by qRTPCR.
Western blot analysis and Antibodies
For western blot analysis, lysed in lysis buffer (50 mM Tris (pH7.4), 150 nM NaCl, 1% NP-40 and 1 mM EDTA, protease/phosphatase inhibitors cocktail (5872S, Cell signaling, Massachusetts, USA)) on ice for 30 min and centrifuged at 15,000 rpm for 15 min. Cell lysates were heat denatured protein, and separated on 8 – 12% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (1620177, Bio-rad, California, USA). Subsequently, blocked with 5% non-fat dry milk (SKI400, Bioshop, Burlington, Canada) and probed with the appropriate primary antibodies.
Antibodies for TLR4 (sc-293072), HDAC2 (sc-9959), β-actin (sc47778) and p16INK4A (sc-56330) were purchased from Santa Cruz (Northern California, USA). SOX9 (82630), H4K8ac (2594) and caspase-3 (9662) antibodies were purchased from Cell signaling technology. p-SOX9 (ab59252) antibody was purchased from Abcam (Cambridge, UK). The bound antibodies were visualized with a mouse (S7076, Cell signaling technology) or a rabbit secondary antibody (S7074, Cell signaling technology) for 1 h and chemiluminescence signals were detected by Clarity ECL reagent (1705060, Bio-rad).
Senescence associated (SA) β-gal activity
Cell staining using senescence β-galactosidase cell staining kit (9860, Cell signaling technology) according to the manufacturer’s instructions. After washing the cells with PBS, replace the 1 X fixative solution for 10 min and incubated at room temperature. Gently wash the cells with PBS, add SA-β-gal staining solution to each well and incubate overnight in a dry 37 ℃ incubator without CO2. Terminate the reaction, observe the cells using microscope (BX-UCB, Olympus, Tokyo, Japan) and represent the SA-β-gal positive cells compared to the total cell number.
Reverse transcription-quantitative PCR analysis
Total RNA was isolated using a RNeasy mini kit (217004, QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Collect the cells through scrapping using cell scraper (90020, SPL, Pocheon-si, Republic of Korea). To lysis the cells, properly add the Buffer RLT. Centrifuge max speed and move the lysate to column. Add 1 volume of 70% ethanol to the lysate and gently pipetting. Transfer 700 μl lysate to RNeasy spin column, incubate for 5 min and centrifuge. Add RW1 buffer to spin column, centrifuge and discard the flow-through. Add RPE buffer and repeat pre-step. Transfer to new collection tube and elute the RNA using RNase free water. Total RNA was reverse transcribed with iScript cDNA synthesis kit (1708891, Bio-rad). 1 μg RNA, 4 μl of 5 x iScript reaction mix, 1 μl iScript reverse transcriptase and RNase free water were added into a 20 μl reverse transcriptase reaction. The reaction was completed under the following conditions: 25 °C for 5 min, 46 °C for 20 min, 95 °C for 1 min. Real-time PCR was performed using an iQ supermix SYBR Green (BR1708884, Biorad). cDNA and specific primer were added into the qPCR Mix and destilled water was added to a total volume of 10 μl. Real-time PCR was performed using a PCR thermal cycler (Thermo Fisher). Real-time PCR was performed using Rotor-Gene Q and software v2.3.1 (QIAGEN). The used primers were described as follows the (Table S1).
|
Name of gene |
Direction |
Nucleotide sequence |
|
HDAC2 |
Forward |
5’-CTC ATG CAC CTG GTG TCC AGA T-3’ |
|
Reverse |
5’-GCT ATC CGC TTG TCT GAT GCT C-3’ |
|
|
Mir-223-3p |
Forward |
5’-TGG GGT ATT TTG ACA AAC TGA CA-3’ |
|
TLR4 |
Forward |
5’-CCC TGA GGC ATT TAG GCA GCT A-3’ |
|
Reverse |
5’-AGG TAG AGA GGT GGC TTA GGC T-3’ |
|
|
p16INK4a |
Forward |
5’-CGC TAA GTG CTC GGA GTT AAT A-3’ |
|
Reverse |
5’-CCG TAA CTA TTC GGT GCG TT-3’ |
|
|
GAPDH |
Forward |
5’-GCT TCC TCT GAC TTC AAC AGC G-3’ |
|
Reverse |
5’-ACC ACC CTG TTG CTG TAG CCA A-3’ |
|
|
U6 |
Forward |
5’-TCGCTTCGGCAGCACATATAC-3’ |
|
Reverse |
5’-TATGGAACGCTTCACGAATTTG-3’ |
|
|
IL-1α |
Forward |
5’-TGTATGTGACTGCCCAAGATGAAG-3’ |
|
Reverse |
5’-AGAGGAGGTTGGTCTCACTACC-3’ |
|
|
IL-1β |
Forward |
5’-CCA CAG ACC TTC CAG GAG AAT G-3’ |
|
Reverse |
5’-GTG CAG TTC AGT GAT CGT ACA GG-3’ |
|
|
IL-6 |
Forward |
5’-AGACAGCCACTCACCTCTTCAG-3’ |
|
Reverse |
5’-TTCTGCCAGTGCCTCTTTGCTG-3’ |
|
|
IL-8 |
Forward |
5’-GAG AGT GAT TGA GAG TGG ACC AC-3’ |
|
Reverse |
5’-CAC AAC CCT CTG CAC CCA GTT T-3’ |
|
|
MCP-1 |
Forward |
5’-AGAATCACCAGCAGCAAGTGTCC-3’ |
|
Reverse |
5’-TCCTGAACCCACTTCTGCTTGG-3’ |
|
|
MCP-2 |
Forward |
5’-TATCCAGAGGCTGGAGAGCTAC-3’ |
|
Reverse |
5’-TGGAATCCCTGACCCATCTCTC-3’ |
|
|
GM-CSF |
Forward |
5’-GGAGCATGTGAATGCCATCCAG-3’ |
|
Reverse |
5’-CTGGAGGTCAAACATTTCTGAGAT-3’ |
Table S1: Primer list for Reverse transcription-quantitative PCR.
Mirco-RNA mimic and inhibition
The miRNA mimics used in this study were as follows: miR-Con-mimic (4464058, Thermo Fisher), miR-223-3p-mimic (4464066, Thermo Fisher), anti-miR-Con-mimic (AM17010, Ambion, Texas, USA) and anti-miR-223-3p (AM17000, Ambion). The sequences used as blow: miR-223-3p mimic: 5′-UGUCAGUUUGUCAAAUACCCCA-3′; anti–miR-223-3p mimic: 5′- UGGGGUAUUUGACAAACUGACA-3′. MiR-223-mimic, anti-miR-223-mimic or control were transfected into HUVECs according to the instruction of Fugene HD Reagent (E2311, Promega, Wisconsin, USA). 48 h after the transfection, expression level of mir-223-3p were measured by RT-PCR.
Cell proliferation assay
The cell proliferation assay by cell counting for 10 days. First, cells are prepared by treating siRNA or reagents according to the prescribed conditions, and then seeded with 2 x 104 cells per well in 6-well plate (30006, SPL). Every 2 days thereafter, the cells are measured for the number in the same volume using the counting chamber. The medium was exchanged every 3 days. The experiment was repeated three times independently.
Flow cytometry
For cell cycle analysis, cells were fixed in 70% ethanol at 4˚C for at least 24 h and incubated in 0.1% of RNase for 30 min at 37˚C. To start analysis, cells were stained with 50 µg/ml of propidium iodide (P3566, Invitrogen) for 30 min at room temperature. Analyses were performed using a BD FACSLyric™ instrument (BD Biosciences, New Jersey, USA). Data obtained from the cell cycle distributions were analyzed with associated software, to determine cell cycle phase distribution.
Fractionation assay
Cytoplasm and nucleus fractionation assay was performed using cell fractionation kit (9038, Cell signaling) according to the manufacturer’s instructions. Briefly, the siRNA or CSE treated cells were washed three times with PBS, followed by CERⅠ buffer 200 µl and incubation for 10 min in ice. Add CER 11 µl with 15 sec of vortex and incubate for 1 min in ice. Centrifuge to 16000 g for 5 min, then obtain the supernatant, which is cytoplasmic extract. And NER 50 µl was added to the remaining pellets and incubated for 40 min in ice after 15 sec of vortex. Centrifuge to 16000 g for 10 min, then obtain the supernatant, which is nucleus extract. The western blotting is carried out with the obtained cytoplasmic and nucleus extract.
Immunofluorescence
The siRNA or CSE treated cells were washed three times with PBS, adding 4% formaldehyde for fixation and incubate for 10 min. After washing three times, incubate with 0.1% IGEPAL (3% BSA in PBS) for 1 h. After washing three times, incubate with SOX9 antibody (1:100 diluted in 3% BSA in PBS) at 4 ˚C overnight. After washing three times, the secondary antibody (Alexa, A11011, 1:100 diluted in 3% BSA in PBS) was incubated at 4 ˚C for 2 h. After washing three times, add DAPI (Cell signaling, 1:1000 diluted in 3% BSA in PBS) and incubate at room temperature for 10 min. At last, the immunofluorescence images were obtained by inverted fluorescence microscope (Olympus, BX-UCB).
Promoter activity assay
The luciferase reporter plasmid of miR-223-3p promoter was cloned into pGL3-basic vector (212936, Addgene). The miR223-3p promoter region was amplified using the follow primers: Forward (5’- GATCG GGTACC GCT TGT GGG TTC AGT-3’) and reverse (5’-GATCG CTCGAG CCA AGA GCT TCT GTG G-3’). Amplified PCR product and vector were digested by Kpn1 and Xho1 for 1 h at room temperature. The ligation was performed using T4 ligase (M0202S, NEB, Nebraska, USA) and confirmed by sequencing service (Macrogen, Seoul, Republic of Korea).
293T cells were seeded 1 x 105 / well in 24-well plate. The miR223-3p promoter-pGL3 basal vector was transfected into 293T cells, and cells treated with or without 5% CSE were obtained, respectively. Luciferase assay was performed using dual-luciferase system (E1980, Promega) according to the manufacturer’s instructions. Briefly, the cells were washed twice and 100 μl of passive lysis buffer was treated well. After incubating for 15 min on orbital shaker with gentle shaking at room temperature, the lysate was moved to 96-well plate each 20 μl per each well. 100 μl of Luciferase Assay Reagent II was added into each well and measured firefly luciferase activity using SpectraMax iD3 (Molecular devices, San Jose, USA). And then, 100 μl of Stop & Glo reagent was added into each well and measured Renilla luciferase activity. Relative luciferase activity was normalized to values obtained using pRL Renilla luciferase (E2231, Promega) as control plasmid. The experiment was repeated three times independently.
Data mining and analysis
We obtained the mRNA and miRNA expression level database from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih. gov/geo/). The GEO database number is GSE 38974. The data platform was Agilent-014850 Whole Human Genome Microarray and miRCURY LNA microRNA Array. The heatmap, expression comparison and correlation graph visualized using GraphPad prism® version 7.01.
The predicted list of transcription factors binding to the miR223-3p promoter was gained from the website below; https:// maayanlab.cloud/Harmonizome. The top 20 lists analyzed by ‘CHEA Transcription Factor Binding Site Profiles’ and ‘JASPAR Predicted Transcription Factor Targets’ were used to select overlapping transcription factors.
Statistics
All experiments were performed in triplicate, and data are shown as means ± SEM. When only two groups were compared, statistical differences were assessed with unpaired a two-tailed Student’s t-test. Otherwise, statistical significance was determined using two-way ANOVA analysis of variance, followed by Bonferroni multiple comparison test. P < 0.05 was considered statistically significant.
Data availability
The GSE datasets gained by GEO profiles section in https://www. ncbi.nlm.nih.gov (Public on Jun 27, 2012). The data generated in the present study may be requested from the corresponding author. The predicted list of transcription factors binding to the promoter was obtained from the website (https://maayanlab.cloud/ Harmonizome).
Funding
This work was supported by National Research Foundation (NRF) of Korea (grant no. NRF-2021R1C1C1006516 and grant no. RS2024-00462318) and Technology Innovation Program (grant no. 20022828; Research and Development of micronized human acellular dermal matrix preserving collagen and growth factor for soft tissue filling) from the Ministry of Trade, Industry & Energy (MOTIE, Korea).
References
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, et al. (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 444: 638-642.
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (2010) The essence of senescence. Genes & development. 24: 2463-2479.
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, et al. (2010) Feedback between p21 and reactive oxygen production is necessary for cell senescence. Molecular systems biology. 6: 347.
- McConnell BB, Starborg M, Brookes S, Peters G (1998) Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Current biology. 8: 351-354.
- Rovillain E, Mansfield L, Lord CJ, Ashworth A, Jat PS (2011) An RNA interference screen for identifying downstream effectors of the p53 and pRB tumour suppressor pathways involved in senescence. BMC genomics. 12: 1-12.
- Coppe JP, Desprez PY, Krtolica A, Campisi J (2010) The senescenceassociated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 5: 99-118.
- Lopes-Paciencia S, Saint-Germain E, Rowell M-C, Ruiz AF, Kalegari P, et al. (2019) The senescence-associated secretory phenotype and its regulation. Cytokine. 117: 15-22.
- Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, et al. (2019) Cellular Senescence: Defining a Path Forward. Cell. 179: 813827.
- Lee BY, Han JA, Im JS, Morrone A, Johung K, et al. (2006) Senescence‐associated β‐galactosidase is lysosomal β‐galactosidase. Aging cell. 5: 187-195.
- van Deursen JM (2014) The role of senescent cells in ageing. Nature. 509: 439-446.
- Vaure C, Liu Y (2014) A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. 5: 316.
- Kim HJ, Kim H, Lee JH, Hwangbo C (2023) Toll-like receptor 4 (TLR4): new insight immune and aging. Immun Ageing. 20: 67.
- Zhang X, Shan P, Qureshi S, Homer R, Medzhitov R, et al. (2005) Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J Immunol. 175: 4834-4838.
- Takyar S, Zhang Y, Haslip M, Jin L, Shan P, et al. (2016) An endothelial TLR4-VEGFR2 pathway mediates lung protection against oxidantinduced injury. FASEB J. 30: 1317-1327.
- Zhang X, Shan P, Jiang G, Cohn L, Lee PJ (2006) Toll-like receptor 4 deficiency causes pulmonary emphysema. J Clin Invest. 116: 30503059.
- Kim SJ, Shan P, Hwangbo C, Zhang Y, Min JN, et al. (2019) Endothelial toll-like receptor 4 maintains lung integrity via epigenetic suppression of p16(INK4a). Aging Cell. 18: e12914.
- Gubbay J, Koopman P, Collignon J, Burgoyne P, Lovell-Badge R (1990) Normal structure and expression of Zfy genes in XY female mice mutant in Tdy. Development. 109: 647-653.
- Rockich BE, Hrycaj SM, Shih HP, Nagy MS, Ferguson MA, et al. (2013) Sox9 plays multiple roles in the lung epithelium during branching morphogenesis. Proc Natl Acad Sci U S A. 110: E4456-64.
- Wang M, Ma Q (2022) Diagnostic biomarkers for skin aging.
- Faleeva M, Ahmad S, Theofilatos K, Lynham S, Watson G, et al. (2024) Sox9 Accelerates Vascular Aging by Regulating Extracellular Matrix Composition and Stiffness. Circ Res. 134: 307-324.
- Inukai S, Slack F (2013) MicroRNAs and the genetic network in aging. J Mol Biol. 425: 3601-3608.
- Izzotti A, Calin GA, Steele VE, Croce CM, De Flora S (2009) Relationships of microRNA expression in mouse lung with age and exposure to cigarette smoke and light. FASEB J. 23: 3243-3250.
- Zhou F, Onizawa S, Nagai A, Aoshiba K (2011) Epithelial cell senescence impairs repair process and exacerbates inflammation after airway injury. Respir Res. 12: 78.
- Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, et al. (2008) Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 451: 1125-1129.
- Mercado N, Ito K, Barnes PJ (2015) Accelerated ageing of the lung in COPD: new concepts. Thorax. 70: 482-489.
- Fukuchi Y (2009) The aging lung and chronic obstructive pulmonary disease: similarity and difference. Proc Am Thorac Soc. 6: 570-572.
- Nyunoya T, Monick MM, Klingelhutz AL, Glaser H, Cagley JR, et al. (2009) Cigarette smoke induces cellular senescence via Werner’s syndrome protein down-regulation. Am J Respir Crit Care Med. 179: 279-287.
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, et al. (2011) Clearance of p16Ink4a-positive senescent cells delays ageingassociated disorders. Nature. 479: 232-476.
- Cheng XY, Li YY, Huang C, Li J, Yao HW (2017) AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget. 8: 22513-22523.
- Boyer L, Chouaid C, Bastuji-Garin S, Marcos E, Margarit L, et al. (2015) Aging-related systemic manifestations in COPD patients and cigarette smokers. PLoS One. 10: e0121539.
- Ito K, Ito M, Elliott WM, Cosio B, Caramori G, et al. (2005) Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 352: 1967-1976.
- Spange S, Wagner T, Heinzel T, Krämer OH (2009) Acetylation of nonhistone proteins modulates cellular signalling at multiple levels. The international journal of biochemistry & cell biology. 41: 185-198.
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 370: 737-749.
- Mizuno S, Yasuo M, Bogaard HJ, Kraskauskas D, Natarajan R, et al. (2011) Inhibition of histone deacetylase causes emphysema. Am J Physiol Lung Cell Mol Physiol. 300: L402-13.
- Wagner M, Brosch G, Zwerschke W, Seto E, Loidl P, Jansen-Durr P (2001) Histone deacetylases in replicative senescence: evidence for a senescence-specific form of HDAC-2. FEBS Lett. 499: 101-106.
- Noh JH, Jung KH, Kim JK, Eun JW, Bae HJ, et al. (2011) Aberrant regulation of HDAC2 mediates proliferation of hepatocellular carcino-ma cells by deregulating expression of G1/S cell cycle proteins. PLoS One. 6: e28103.
- Li S, Wang F, Qu Y, Chen X, Gao M, et al. (2017) HDAC2 regulates cell proliferation, cell cycle progression and cell apoptosis in esophageal squamous cell carcinoma EC9706 cells. Oncol Lett. 13: 403-409.
- Wong QWL, Lung RWM, Law PTY, Lai PBS, Chan KYY, et al. (2008) MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology. 135: 257269.
- Vallelunga A, Ragusa M, Di Mauro S, Iannitti T, Pilleri M, et al. (2014) Identification of circulating microRNAs for the differential diagnosis of Parkinson’s disease and Multiple System Atrophy. Front Cell Neurosci. 8: 156.
- Leuenberger C, Schuoler C, Bye H, Mignan C, Rechsteiner T, et al. (2016) MicroRNA-223 controls the expression of histone deacetylase 2: a novel axis in COPD. J Mol Med (Berl). 94: 725-734.
- Booth LN, Brunet A (2016) The Aging Epigenome. Mol Cell. 62: 728744.
- Vilas JM, Carneiro C, Da Silva-Alvarez S, Ferreiros A, Gonzalez P, et al. (2018) Adult Sox2+ stem cell exhaustion in mice results in cellular senescence and premature aging. Aging Cell. 17: e12834.
- Foronda M, Martinez P, Schoeftner S, Gomez-Lopez G, Schneider R, et al. (2014) Sox4 links tumor suppression to accelerated aging in mice by modulating stem cell activation. Cell Rep. 8: 487-500.
- Tchougounova E, Jiang Y, Bråsäter D, Lindberg N, Kastemar M, et al. (2009) Sox5 can suppress platelet-derived growth factor B-induced glioma development in Ink4a-deficient mice through induction of acute cellular senescence. Oncogene. 28: 1537-1548.
- Matheu A, Collado M, Wise C, Manterola L, Cekaite L, et al. (2012) Oncogenicity of the developmental transcription factor Sox9. Cancer Res. 72: 1301-1315.
- Stevanovic M, Lazic A, Schwirtlich M, Stanisavljevic Ninkovic D (2023) The role of SOX transcription factors in ageing and age-related diseases. International Journal of Molecular Sciences. 24: 851.
- Rui L, Liu R, Jiang H, Liu K (2022) Sox9 Promotes Cardiomyocyte Apoptosis After Acute Myocardial Infarction by Promoting miR-223-3p and Inhibiting MEF2C. Mol Biotechnol. 64: 902-913.
- Zhu X, Huang H, Zong Y, Zhang L (2022) SRY-related high-mobility group box 9 (SOX9) alleviates cigarette smoke extract (CSE)-induced inflammatory injury in human bronchial epithelial cells by suppressing stromal interaction molecule 1 (STIM1) expression. Inflamm Res. 71: 565-576.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.