Vitamin D as a Renoprotective Remedy Against Non-Proteinuric Diabetic Kidney Disease
by Michael Richards, Sydney Lance, Mohamed Ahmed*
California Northstate University, College of Medicine, West Taron Drive, Elk grove, CA 95757, USA
*Corresponding author: Mohamed Ahmed, MD, MSc Assistant Professor of Anatomy and Embryology, Department of Basic Sciences, College of Medicine, California Northstate University, West Taron Drive, Elk Grove, CA 95757 USA
Received Date: 08 September 2025
Accepted Date: 15 September 2025
Published Date: 17 September 2025
Citation: Richards M, Lance S, Ahmed M (2025) Vitamin D as A Renoprotective Remedy Against Non-Proteinuric Diabetic Kidney Disease. J Urol Ren Dis 09: 1434. https://doi.org/10.29011/2575-7903.001434
Abstract
Diabetic Kidney Disease (DKD) is a leading cause of Chronic Kidney Disease (CKD) and end-stage renal disease worldwide, significantly contributing to morbidity and mortality. Among its forms, non-proteinuric DKD is increasingly recognized as a distinct clinical entity characterized by renal dysfunction without the hallmark proteinuria seen in traditional diabetic nephropathy. This literature review aims to evaluate the potential renoprotective role of vitamin D in non-proteinuric DKD and synthesizes current evidence on vitamin D’s mechanisms of action, including regulating pro-inflammatory cytokines such as IL-6 and TNF-α, inhibiting RAAS hyperactivation, mineral homeostasis, and reducing fibrosis through modulation of TGFβ and connective tissue growth factors. Additionally, it highlights vitamin D as a promising adjunctive therapy for non-proteinuric DKD, warranting further investigation to establish its clinical utility and integration into comprehensive management strategies. Furthermore, vitamin D’s potential synergy with established renoprotective agents, such as Angiotensin-Converting Enzyme Inhibitors (ACEis), Angiotensin II Receptor Blockers (ARBs), and Sodium-Glucose Cotransporter 2 (SGLT2) inhibitors, is explored. While preclinical and clinical studies suggest that vitamin D supplementation may mitigate inflammation and fibrosis in DKD, conflicting evidence exists regarding its efficacy in improving renal function metrics such as Glomerular Filtration Rate (GFR) in non-proteinuric cases. Gaps in research, particularly regarding optimal dosing and long-term outcomes, underscore the need for randomized controlled trials in diverse populations.
Keywords: Bone-Mineral Disorder; Chronic Kidney Disease; Diabetic Kidney Disease; Non-Proteinuric Kidney Disease; Renoprotection; Vitamin D
Methodology
A systematic literature search (PubMed, Embase, Cochrane Library, CNKI, WANGFANG, and VIP) was performed to identify articles published between September 2007 and July 2018. The search items were vitamin D, cholecalciferol, calcitriol, 1, 25(OH) D3, paricalcitol, and diabetic nephropathy, diabetes, chronic kidney disease in the title, abstract, and keywords, with no restriction imposed. Additional papers were found through a manual search for reference lists of review articles.
Chronic Kidney Disease
Chronic Kidney Disease (CKD) is defined as an abnormal kidney structure or function that lasts more than 3 months [1]. To be diagnosed with CKD, the patient must have decreased Glomerular Filtration Rate (GFR) {<60mL/min per 1.73 m2} alone or decreased GFR and another marker of kidney damage, including albuminuria (Albumin: Creatinine Ratio ≥30mg/g), hematuria, abnormal structure as determined by imaging, abnormal histology, history of a kidney transplant, and urine sediment abnormalities [1]. CKD can be caused by a primary kidney disease or a systemic disease affecting the kidneys. The pathophysiology of CKD will vary depending on the cause. Worldwide, the most common causes of CKD are diabetes and hypertension [1]. CKD is categorized using the CGA classification for Cause, GFR, and Albuminuria [1]. GFR is divided into categories G1-G5, with G1 being a GFR ≥90 and G5 being kidney failure or a GFR <15. Albuminuria is divided into A1-A3, with A1 being normal to mildly increased albuminuria and A3 being severely increased at >30 mg/g. Higher stages of CKD are linked to poorer outcomes [2]. Patients are often asymptomatic until advanced disease stages, at which point they can develop elevated blood pressure, anemia, dyslipidemia, bone disease, electrolyte abnormalities, among other symptoms [1]. This highlights the importance of screening, especially in patients with risk factors like diabetes [3,4] (Figure 1).
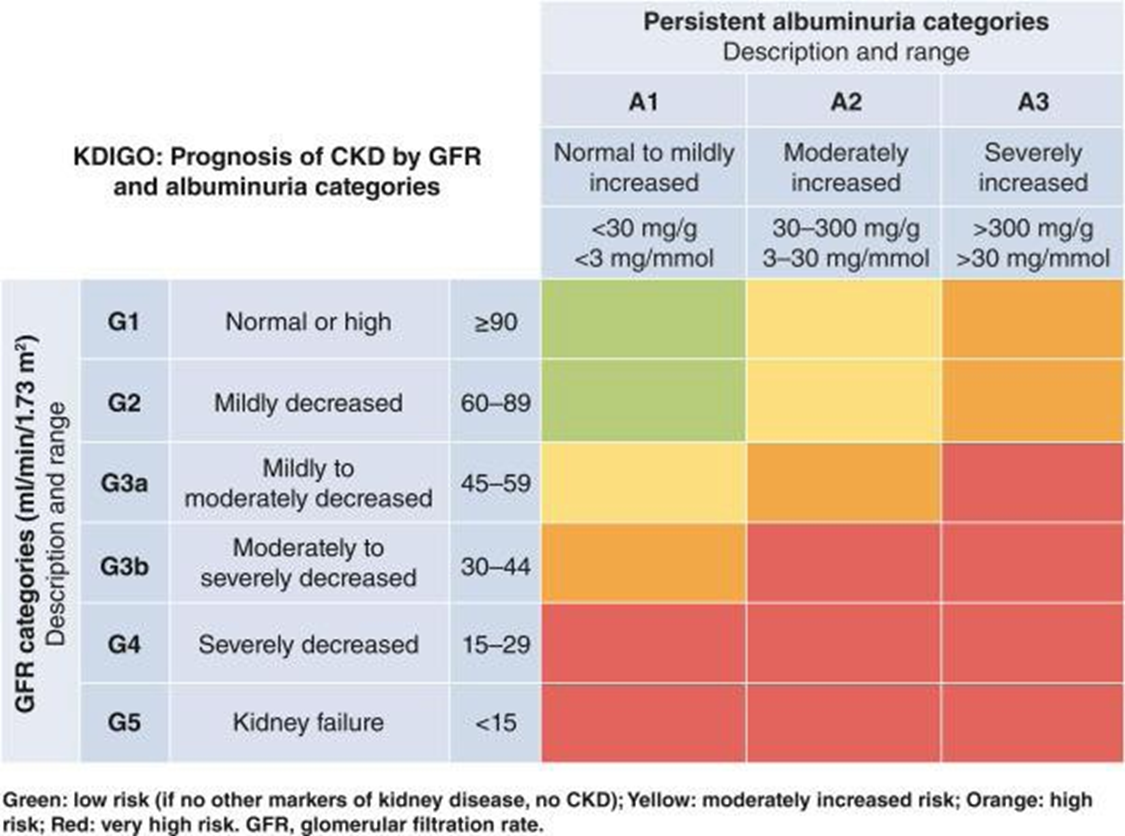
Figure 1: Categorization and prognosis of CKD by GFR and albuminuria.Pathophysiology of CKD. 2023 kidney disease: Improving Global Outcomes (KDIGO) https://doi.org/10.1016/j.kint.2023.10.018.
In response to a stressor such as hypertension or diabetes, parenchymal cells and infiltrating immune cells release growth factors, reactive oxygen species, and other effector molecules that cause proliferation of myofibroblasts and mesangial cells, as well as injury to podocytes, the resulting fibrosis and glomerulosclerosis lead to CKD (Figure 2).
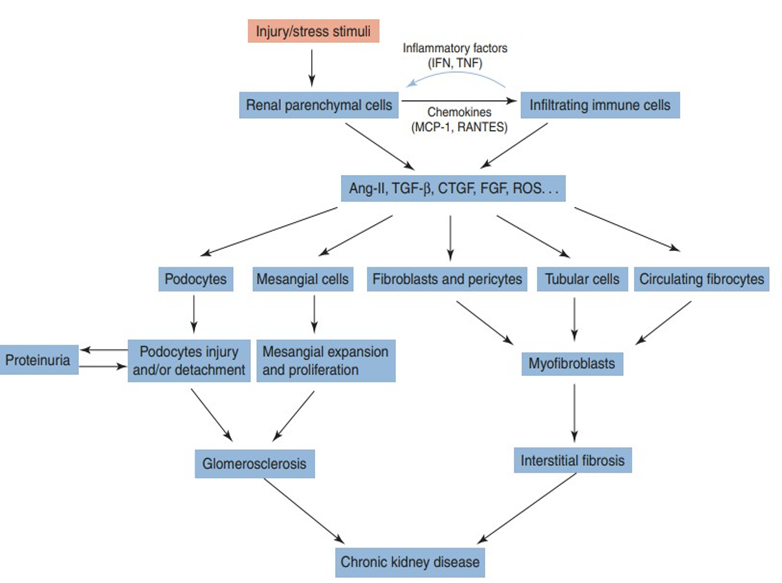
Figure 2: Schematic presentation of cellular events involved in the progression of chronic kidney disease. J. Yang, W. He (eds.), chronic kidney disease.
Diabetic Kidney Disease
Diabetic Kidney Disease (DKD) is the most common cause of CKD in the United States [1]. DKD is a major complication of diabetes, occurring in up to 50% diabetic patients and contributing significantly to morbidity and mortality. Given the growing prevalence of diabetes and its complications, exploring effective therapeutic strategies, including vitamin D supplementation, is imperative (Table 1).
Prevalence of diabetic vs. nondiabetic kidney disease
Table 1: Prevalence (95% confidence interval) of diabetic versus nondiabetic kidney disease versus in the US population, NHANES 2009–2014.
|
CKD |
UACR≥300 mg/g |
EGFR <60 ml/min per 1.73 m2 |
|
|
Diabetes, % |
25 (21 to 28) |
4.6 (3.4 to 5.8) |
12 (9 to 15) |
|
No biabetes, % |
5.3 (4.6 to 5.9) |
0.3 (0.2 to 0.5) |
2.5 (2.0 to 3.0) |
Data are adopted from Zelnick et al. [Zelnick LR, Weiss NS, Kestenbaum BR, Robinson-Cohen C, Heagerty PJ, Tuttle K, Hall YN, Hirsch IB, de Boer IH. Diabetes and CKD in the United States population, 2009-2014. Clin J Am Soc Nephrol. 2017;12(12):1984–90]
Pathophysiology of DKD
The pathophysiology of DKD involves several interrelated mechanisms contributing to renal damage and functional decline. The primary factors involved include chronic hyperglycemia, RAAS activation, inflammation and fibrosis, altered hemodynamics, mitochondrial dysfunction, and oxidative stress (Figure 3).
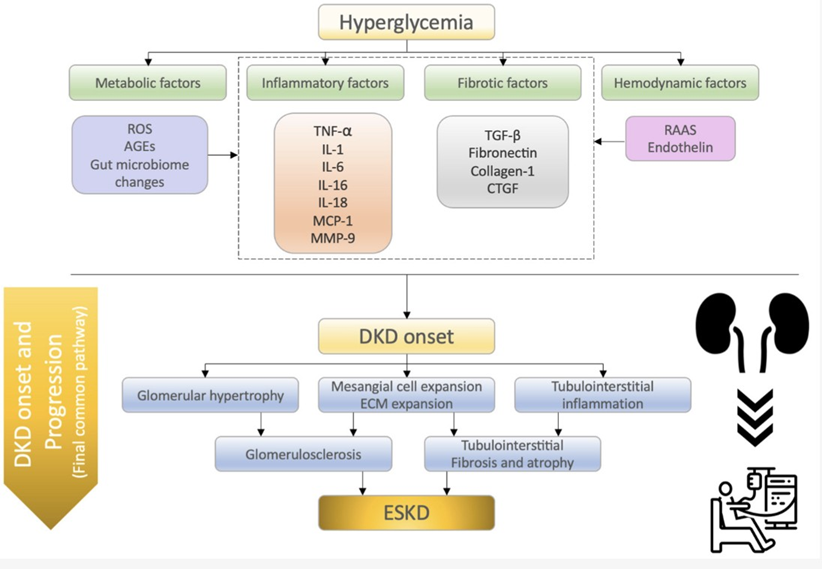
Figure 3: Summary of the molecular mechanisms of diabetic kidney disease (DKD) onset and progression. Watanabe, K. et al. What’s new in the molecular advances of diabetic kidney disease: Recent advances.
Inflammation and Fibrosis in DKD
Persistent inflammation from attempts to repair renal damage leads to the upregulation of many cytokines and growth factors that eventually lead to fibrosis and diminish renal function over time [5,6]. Interleukin-6 (IL-6) is a critical inflammatory cytokine as it plays a role in both early and late inflammation and can be used as a biomarker for the progression of DKD [7]. Early in the inflammatory state, IL-6 promotes the removal of neutrophils and allows for the infiltration of macrophages [8]. These macrophages can be of the classically activated M1 phenotype, which releases other pro-inflammatory cytokines such as interleukin- 1ß (IL1β) and tumor necrosis factor-α (TNFα), or the alternatively activated M2 phenotype, which is anti-inflammatory and strongly associated with fibrosis [9]. IL-6 plays a role in the balance of M1 and M2 macrophages by inhibiting the M1 phenotype late in the inflammatory process, and this switch from predominant M1 to M2 macrophages marks the transition from inflammation to chronic fibrosis [8,9]. M2 macrophages both release and are activated by transforming growth factor- ß (TGFβ). TGFβ is significant in fibrosis because it induces the transformation of other cell types, such as fibroblasts and pericytes, into myofibroblasts, which are responsible for laying the extracellular matrix (ECM) that eventually becomes fibrotic scar tissue [10]. TGFβ also induces the synthesis of connective tissue growth factor (CTGF), another profibrotic growth factor that can induce certain cells to become myofibroblasts [10]. CTGF also induces the production of collagen and fibronectin, as well as the important inflammatory mediator IL-6 [11]. These inflammatory and fibrotic processes are intrinsically linked to the mitochondrial dysfunction seen in DKD [12]. Hyperglycemia leads to enhanced mitochondrial oxidative phosphorylation, subsequently resulting in increased Reactive Oxygen Species (ROS) production, which can damage cellular components and cause an inflammatory response governed by TGFβ and IL-6 [13]. This inflammatory environment contributes to further mitochondrial dysfunction and creates a feedback loop that accelerates renal injury [14,15]. Prolonged oxidative stress and inflammation activate fibroblasts and stimulate ECM deposition, leading to renal tissue scarring [16].
- Chronic Hyperglycemia causes an accumulation of Advanced Glycation End Products (AGEs), which are formed via non-enzymatic glycation of proteins and lipids [17]. The byproducts of these irreversible reactions are permanently deposited throughout the body, and act via several pathways, including the receptor for AGEs (RAGE), resulting in renal cell dysfunction and apoptosis, oxidative damage, and inflammation [18]. Podocytes and endothelial cells are particularly at risk of damage [15]. Podocyte injury is more prominent in the proteinuric form, where damage to podocytes from AGEs and oxidative stress results in loss of the glomerular filtration barrier, causing protein leakage in the urine [14]. Specifically, podocytes are lost, and the remaining ones are required to grow and extend their foot processes in order to cover the same surface area. As a result, there are fewer and less effective podocytes present in the filtration barrier, causing a decline in kidney function and eventual sclerosis [19]. This level of podocyte damage is not seen in the non-proteinuric form, leaving the filtration barrier intact despite significant renal cell damage. Mouse studies have also implicated hyperglycemia in the overexpression of TGFβ in glomerular mesangial cells, and subsequent fibrosis [20]. This combination of fibrosis and inflammation causes further damage to glomerular cells, leading to progressive renal dysfunction.
- Renin-Angiotensin-Aldosterone System (RAAS) is often overactive, leading to increased production of angiotensin II, which contributes to glomerular hypertension and hyperfiltration [21]. In renal cells, hyperglycemia and AGEs directly induce the expression of renin and angiotensin via the production of Reactive Oxygen Species (ROS) and other mediators [21]. Angiotensin is then converted to Angiotensin II, which promotes vasoconstriction of the afferent arterioles and stimulates the secretion of aldosterone, resulting in sodium retention, further exacerbating hypertension and kidney damage [22].Aldosterone also regulates gene expression for the pro-sclerotic factors plasminogen activator inhibitor 1 (PAI-1) and TGFβ and promotes macrophage infiltration [21]. Both processes accelerate the development of inflammation and fibrosis, leading to the progression of DKD.
- Oxidative Stress: Hyperglycemia may cause oxidative stress, or the accumulation of ROS. These ROS stimulate the protein kinase B, uncoupling protein 2 (UCP2) and JAK-STAT pathways, which result in proliferation of glomerular mesangial cells and increased expression of matrix proteins [23]. This causes increased fibrosis in glomerular cells. Chronic hyperglycemia is associated with decreased superoxide dismutase [24] and increased malondialdehyde [23], which are markers of oxidative stress. Oxidative stress stimulates several pro-inflammatory and profibrotic pathways that lead to, extracellular membrane deposition, podocyte damage, thickened glomerular basement membrane and mitochondrial damage, all of which contribute to decline in eGFR [25].
- Hemodynamics: In addition to glomerular damage due to overactive RAAS, altered hemodynamics in diabetes can directly cause renal damage. The inflammation and endothelial damage from hyperglycemia lead to vascular stiffening and vasoconstriction, which may cause hyperfiltration while also decreasing perfusion to the kidney, resulting in renal damage [26]. It has been shown in clinical trials that elevated blood pressure is a risk factor for end-stage renal disease and is associated with a faster decline in eGFR [27,28].
Subdivisions of Diabetic Kidney Disease
Diabetic Kidney Disease (DKD) is a multifactorial complication of diabetes that results from prolonged hyperglycemia and its associated metabolic disturbances. While persistent proteinuria has been considered a cardinal feature of DKD for some time [15], it has been found that a significant portion of diabetics experience renal dysfunction without proteinuria [16] and that most patients with diabetes and CKD have no albuminuria at all [29]. In the Renal Insufficiency and Cardiovascular Events (RIACE) trial, 57% of patients with type 2 diabetes with CKD had normoalbuminuria [30], and other cross-sectional studies reported similar incidence of normoalbuminuria in DKD patients including the National Evaluation of the Frequency of Renal Impairment Coexisting with type 2 Diabetes (NEFRON) study reporting 55% normoalbuminuria [31], and 40% in the Developing Education on Microalbuminuria for Awareness of Renal and Cardiovascular Risk in Diabetes (DEMAND) study [32]. Additionally, it has been found that patients with non-proteinuric DKD can transition to the proteinuric form over time [8].
• Proteinuric DKD
Proteinuric DKD (PDKD) follows a classic progression of initial glomerular hyperfiltration, followed by proteinuria and a gradual decrease in the GFR and microalbuminuria that may lead to end-stage kidney disease. Proteinuric DKD is also characterized by abnormal renal pathology. Nodular glomerulosclerosis is the hallmark lesion on biopsy and glomerular basement thickening and hyalinosis lesions [16]. Podocyte injury and death drive many of the changes in proteinuric DKD. Altered hemodynamics in diabetes cause an initial increase in GFR followed by a decline as nephron injury progresses [21]. The hyperfiltration state contributes to further podocyte injury and loss, which is pivotal in developing proteinuric DKD and progressing Non Proteinuric DKD (NPDKD) to the proteinuric form [33]. Reduced energy availability in diabetes also enhances susceptibility to cell injury and death [34]. Mitochondria play a crucial role in calcium signaling, which in proteinuric DKD can further compromise glomerular function, as dysregulated intracellular calcium levels are associated with podocyte apoptosis and cellular fibrosis, exacerbating proteinuria [12]. Podocyte injury is the primary driver of inflammation in the proteinuric form of DKD as injured cells release pro-inflammatory cytokines [35].
Non-Proteinuric DKD
In contrast to the proteinuric form, biopsies of patients with NPDKD often reveal normal renal structure or may lack nodular glomerulosclerosis that characterizes proteinuric DKD [14].
However, a study by Chang et al. showed that, while many NPDKD patients showed evidence of typical glomerular lesions, only 22.2% of these patients displayed the same severity of damage as their matched PDKD counterparts [3]. NPDKD may be associated with relatively preserved glomerular function until the later stages, complicating early diagnosis [16]. This underscores the importance of examining DKD from a broader perspective than just the overt presence of proteinuria. It is possible that the reduced levels of typical lesions in NPDKD is due to the preponderance of macroangiopathic lesions in comparison to the microangiopathic ones typically seen in the proteinuric form [10]. This is further emphasized by the reduced prevalence of retinopathy, a process dominated by microangiopathic damage, in non-proteinuric patients [3]. The RIACE trial showed that non-proteinuric renal impairment was strongly associated with major cardiovascular events, further suggesting a macro-vascular component to the disease [30]. This key difference could be an opportunity for further investigation. In contrast to proteinuric DKD, non-proteinuric DKD reflects a broader inflammatory response that impacts tubular and interstitial compartments of the kidney [14]. These inflammatory changes are critical in determining the potential role of vitamin D as part of a renoprotective strategy, as reduced levels of 1,25-dihydroxyvitamin D are associated with the promotion of renal inflammation and fibrosis [36]. In NPDKD, oxidative stress is primarily a result of impacts on energy production within the mitochondria, resulting in reduced levels of ATP in renal cells, negatively impacting critical cellular functions such as ion transport, maintenance of cell membrane integrity, and synthesis of protective molecules [3]. Renal hemodynamic alterations in the non-proteinuric form are often more independent from the hyperfiltration seen in PDKD [37]. Patients may experience early renal blood flow and GFR declines, which are associated with reduced nitric oxide availability, subsequent vascular stiffness, and impaired renal perfusion [37]. Recent studies have also suggested that non-proteinuric patients are likely to have significantly more atherosclerosis, leading to increased vascular resistance and glomerular-tubular damage [9].
• Possible Mechanism of NPDKD
NPDKD is caused by abnormalities in the vascular or predominantly tubulointerstitial abnormalities [38,39], although it can also be a typical disorder [14,40]. NPDKD pathophysiological investigations should provide additional insight into cardiovascular factors affecting kidney function and disease and provide new treatments for the vascular complications seen in diabetic patients [41]. There are several possibilities for this NPDKD, including the accompanying vascular disease (there is an increase in interlobar artery vascular resistance) [42] which causes damage to glomerular and tubular structures and interstitial fibrosis [43]: the result of previous episodes of Acute Kidney Injury (AKI), which is related to the inherent susceptibility of diabetic patients [44]; the existence of a well-preserved tubule that leads to a significant reabsorption of albumin from the glomerular filtrate, thus resulting in a diminished albumin excretion into normoalbuminuric levels [43]; and an increase in intrarenal arteriosclerosis as opposed to classical glomerulosclerosis changes present in albuminuric subjects [44]. The main contributing factors to progression to ESRD are AKI, as has been proven by Thakar et al. in 2011. In 2019, Sykes et al. found that AKI events were associated with progression to renal replacement rate and with a greater severity of subsequent AKI [45,46]. This may explain why some DN patients have an early decline of GFR with a minimal amount of albuminuria.
Additionally, There are Other Factors That Also Play A Role
(1) Increased levels of uric acid, which can damage vascular elements and cause endothelial dysfunction through various mechanisms, including activation of the Toll-like receptor pathway [47,48]. Uric acid can also induce renal inflammation, proliferation of vascular smooth muscle cells, and activation of the RAS [49-52].
(2) Increased concentration of serum TNF-α [53]. TNF-α is a major mediator of inflammation and is involved in AKI, regulation of blood pressure, blood flow, inflammation in the renal blood vessels [54-56], and apoptosis [53]. Thus, elevated levels of TNF-α can alter renal blood vessels and damage the kidneys.
• Prognosis : NPDKD has a better prognosis than PCKD with significant proteinuria. This is in accordance with the understanding so far, that the degree of proteinuria is a strong predictor of the risk of progression [40]. However, compared with no kidney disease, non-proteinuric diabetic nephropathy (NPDN) was a significant risk factor for death and major cardiovascular disease [57]. Diagnosis of NPDN is based on an increase in the Renal Resistive Index (RRI), which measures renal vascular resistance. RRI measurements have been shown to be reliable for detecting and monitoring the development of diabetic nephropathy (DN) and NPDN [58-60]. A study in diabetic patients showed an increased RRI value in diabetic patients without proteinuria or renal atherosclerosis [61]. Therefore, ultrasound sonography provides an effective method to screen, identify, and monitor hemodynamic and morphological changes in DN patients [61].
Biochemistry of Vitamin D
- Vitamin D metabolism is a crucial process influencing various physiological functions, including calcium homeostasis and immune regulation. It involves several steps and primarily occurs in the skin, liver, and kidneys.
- Synthesis in the Skin: Vitamin D is synthesized in the skin upon exposure to ultraviolet B (UVB) radiation from sunlight. 7-dehydrocholesterol in the skin is converted to previtamin D3, which is subsequently transformed into vitamin D3 (cholecalciferol). This process is influenced by factors such as skin pigmentation, geographic location, and season [62].
- Liver Conversion: Once synthesized or ingested, vitamin D3 is transported to the liver, where it undergoes hydroxylation to form 25-hydroxyvitamin D (25(OH)D or calcidiol) through the action of the enzyme vitamin D 25-hydroxylase. This is the primary circulating form of vitamin D and indicates vitamin D status in the body [63]. 25 (OH)D governs the pathologies of vitamin D, including insufficiency and deficiency, so that this is the analyte that is measured in clinical diagnostic labs [63-65].
- Kidney Conversion: The final activation step occurs in the kidneys, where 25(OH)D is further hydroxylated to form 1,25-dihydroxyvitamin D (1,25(OH)2D or calcitriol), the tightly controlled biologically active form of vitamin D. This process is mediated by the enzyme 1α-hydroxylase, which is regulated by several factors, including parathyroid hormone (PTH), serum calcium, and phosphate levels [64,65].
- 1,25(OH)2D exerts its effects through the Vitamin D Receptor (VDR) expressed in various tissues, including the kidneys. In the kidneys, 1,25(OH)2D enhances calcium reabsorption [63]. It modulates the expression of proteins involved in renal function, including those related to inflammation and fibrosis.
- This reduction in inflammation is likely the most critical role that vitamin D can play in the context of non-proteinuric DKD, as it has been shown to lower levels of inflammatory markers such as hs-CRP, TNF-α, and IL-6 [66]. However, it is essential to note that this meta-analysis did not show a significant influence of vitamin D on serum creatinine levels or eGFR. However, an anti-inflammatory effect can be seen due to negative regulation of the RAAS system. In a 2008 study, Zhang et al. demonstrated that VDR knockout mice developed hyperreninemia resulting in hypertension and cardiac hypertrophy. This study also demonstrated that VDR knockout mice exhibited higher levels of angiotensinogen, TGFβ, and connective tissue growth factor. Additionally, 1,25(OH)2 inhibited fibronectin production in mesangial cells that were stimulated by high glucose levels and increased nephrin expression in podocytes [20]. These findings suggest that vitamin D could potentially mitigate the hypertensive effects of RAAS over-stimulation in diabetic nephropathy and the release of pro-inflammatory and pro-fibrotic cytokines.
Vitamin D and Kidney Health
Vitamin D plays a critical role in calcium and phosphate homeostasis and exerts various effects on renal physiology. Research indicates that vitamin D deficiency is prevalent among patients with CKD, potentially exacerbating renal damage [63,67]. Vitamin D’s protective mechanisms may involve modulation of inflammation, regulation of RAAS activity, and promotion of endothelial function [13,18]. The kidneys, intestines, and parathyroid glands work in concert to maintain calcium and phosphate homeostasis [68]. In response to hypocalcemia, elevated phosphate levels, or low levels of 1,25-dihydroxyvitamin D (calcitriol), the parathyroid glands secrete Parathyroid Hormone (PTH). PTH acts directly on bones to mobilize calcium and phosphate into the bloodstream [69]. In the kidneys, PTH downregulates phosphate transporters in the proximal convoluted tubule (PCT), promoting phosphaturia, and upregulates 1α-hydroxylase to enhance calcitriol production [70]. Increased calcitriol subsequently promotes calcium absorption in the small intestine and reabsorption in the distal renal tubules [71]. Fibroblast growth factor 23 (FGF23), primarily secreted in response to phosphate overload, works alongside PTH to induce phosphaturia [72]. It downregulates type II sodium-dependent phosphate transporters in the renal PCT and inhibits calcitriol synthesis, thereby lowering serum phosphate levels [73]. FGF23 requires α-Klotho as a co-receptor, which ispredominantly expressed in the distal renal tubules, parathyroid glands, and choroid plexus [73]. In CKD, nephron loss leads to reduced α-Klotho expression and impaired FGF23 signaling, contributing to hyperphosphatemia [73]. To maintain phosphate balance, circulating levels of FGF23 and PTH rise, enhancing phosphate excretion [74,75]. However, this compensatory mechanism results in secondary hyperparathyroidism, reduced calcitriol levels, and eventual hypocalcemia-hallmarks of advanced CKD [74]. The kidneys, intestines, and parathyroid glands work closely to maintain calcium and phosphate homeostasis [68]. In response to low calcium levels and 1,25(OH)2 or elevated phosphate, the parathyroids release PTH, which acts directly on bone to release calcium and phosphate into circulation [73,74]. PTH simultaneously downregulates phosphate transporters in the renal PCT cells to induce phosphaturia [70] and upregulates 1a-hydroxylase in the kidneys to increase the production of calcitriol, thereby increasing calcium reabsorption in the small intestine and distal renal tubules [71]. Additionally, Fibroblast growth factor 23 (FGF23) works with PTH to induce phosphaturia [72]. In the setting of phosphate overload, which also induces increased PTH production, FGF23 concentrations increase. FGF 23 downregulates type-II sodiumdependent phosphate transporters in the PCT and deactivates calcitriol to reduce serum phosphate concentrations [72,74]. FGF 23 action requires a-klotho as a co-receptor, which is found primarily in the renal distal tubules, as well as the choroid plexus and parathyroid glands [73]. In the setting of chronic kidney disease of any kind, nephrons are lost, resulting in fewer of these receptors, and subsequent decreased action of FGF 23 and resulting hyperphosphatemia [73]. To compensate, urinary excretion of phosphate must increase, which is accomplished by increasing secretion of FGF23 and PTH [74,75]. This in turn results in secondary hyperparathyroidism often seen in later stage kidney disease, as well as decreased calcitriol concentration and eventual decline in calcium concentration [73,74,75].
Chronic Kidney Disease- Mineral and Bone Disorder (CKDMBD)
The metabolic disruption caused by CKD extends beyond endocrine and mineral abnormalities, culminating in a condition known as chronic kidney disease–mineral and bone disorder (CKD-MBD) [69,76]. CKD-MBD is typically characterized by hypocalcemia and hyperphosphatemia, particularly in the later stages of CKD. However, this dysregulation of mineral homeostasis likely begins earlier in the course of disease, contributing to a cascade of downstream effects [71]. Once thought to be limited to parathyroid and skeletal pathology, CKD-MBD is now recognized as a contributor to the increased cardiovascular morbidity and mortality seen in CKD patients [77]. This is largely due to the deposition of calcium-phosphate crystals in blood vessels and cardiac valves, a process known as cardiovascular calcification. In CKD, this calcification occurs predominantly in arterial walls and heart valves through an actively regulated, cell-mediated process involving Vascular Smooth Muscle Cells (VSMCs), Vascular Interstitial Cells (VICs), macrophages, and endothelial cells [71]. During this process - referred to as osteochondrogenic differentiation - VSMCs and VICs lose their contractile phenotype and adopt a synthetic phenotype resembling osteoblasts [78]. These osteoblast-like cells secrete bone-associated proteins that promote ECM calcification [78,79]. The process is further accelerated in CKD by increased cellular apoptosis, oxidative stress, and mitochondrial dysfunction [80]. Additionally, mineral imbalances such as hyperphosphatemia, hypercalcemia, uremic toxins, and pro-inflammatory cytokines act as potent inducers of vascular calcification [80]. Management of CKD-MBD focuses on controlling PTH levels. According to the Kidney Disease Improving Global Outcomes (KDIGO) guidelines [81], PTH levels in dialysis patients should be maintained at 2–9 times the upper limit of normal. Persistent elevations in PTH may require correction of underlying abnormalities such as hyperphosphatemia, hypocalcemia, and vitamin D deficiency [82]. Treatment with active vitamin D analogs such as calcitriol has been shown to reduce bone turnover markers and lower PTH levels in these patients [83].
Vitamin D Effects on DKD
Vitamin D plays an important role in diabetes outside the realm of DKD. Insulin secretion is dependent on calcium, and there is evidence that vitamin D deficiency can impair insulin secretion [29]. Vitamin D has also been shown to downregulate inflammatory markers such as IL-6 and TNF-α, which are elevated in diabetic patients [30]. Several longitudinal studies have shown an inverse relationship between vitamin D levels and the prevalence of diabetes [29]. Severe vitamin D deficiency in patients with type 2 diabetes is associated with an increase in all-cause mortality [84]. Taken together, these findings suggest a role for Vitamin D in the primary prevention of diabetes, even before kidney damage takes place.In experimental animals, vitamin D deficiency associates with an earlier and more aggressive onset of diabetes, probably related to abnormalities in immune function, and impaired glucose-mediated insulin secretion that can be reversed by calcitriol repletion [85].1,25(OH)2D3, through a VDR-mediated modulation of calbindin expression, appears to control intracellular calcium flux in the islet cells, which, in turn, affects insulin release [86]. In the 1,25(OH)2D3 deficiency of CKD, there is abnormal insulin secretion, a blunted response of the pancreatic cells to glucose challenge, and insulin resistance [87-89]. 1,25(OH)2D3 deficiency produces abnormal regulation of insulin secretion independently of alterations in VDR levels in pancreatic cells. Also, 1,25(OH)2D3 administration corrects the abnormal insulin secretion independently of changes in serum levels of calcium or PTH [90,91]. The finding of 1-hydroxylase activity in pancreatic cells [92] raises the possibility of an autocrine control of insulin secretion by 1,25(OH)2D3.
The half-life of 1,25(OH)2D is relatively short, and its metabolites are primarily excreted via the kidneys [93]. Impaired renal function can reduce the conversion of vitamin D to its active form, contributing to vitamin D deficiency, which is commonly observed in patients with CKD [93]. Several studies have suggested a beneficial association between vitamin D levels and renal outcomes in diabetic patients [66]. For example, a systematic review indicated that vitamin D supplementation may improve renal function and reduce inflammation in patients with DKD [66]. However, conflicting evidence exists regarding the efficacy of vitamin D in the absence of proteinuria, necessitating further investigation into optimal dosing and long-term effects [41].While not directly related to its potential impact on kidney function, there has also been research indicating that Vitamin D plays a significant role in reducing plaque formation in atherosclerosis [65]. Specifically, a study found that macrophages of diabetic patients cultured without Vitamin D would produce more foam cells than those cultured with it [94]. This study also showed that Vitamin D-sufficient macrophages took up less cholesterol than their deficient counterparts. The combination of these factors could result in increased plaque formation and subsequent macroangiopathic complications.
Renoprotective Regimens: ACEis, ARBs, and SGLT2 Inhibitors
In addition to vitamin D, several pharmacological agents have demonstrated renoprotective effects in DKD [22]. ACE inhibitors and ARBs are widely recognized for their ability to lower blood pressure and reduce albuminuria, leading to improved renal outcomes [22]. These medications function by inhibiting (RAAS), which is often overactive in diabetic patients, contributing to glomerular damage. SGLT2 inhibitors have also emerged as a promising class of medications in the management of DKD. Recent studies show that SGLT2 inhibitors can significantly reduce the risk of progression to end-stage kidney disease in patients with diabetes [95]. The renoprotective mechanisms of GFSGLT2 inhibitors include improvements in glycemic control, reductions in hyperfiltration, and beneficial effects on cardiovascular health [96].
Potential mechanism of Vitamin D in the treatment of NPDKD
The most probable mechanism by which Vitamin D can play a role in NPDKD lies in the demonstrated negative regulation of the RAAS system, reduced pro-inflammatory cytokine production, delaying the progression of CKD-MBD, and reduced fibrosis. As has been described by Zhang et al., intrarenal RAAS is increased in patients with diabetic nephropathy and plays a key role in its development [8]. This increase in RAAS activation results in increased levels of angiotensin II, which is directly implicated in increased TGFβ expression and ECM protein production in mesangial and tubular cells [14,15]. This, in combination with the hypertension associated with RAAS activation, directly results in renal injury and inflammation [14,15,16]. This activation level was even more prominent in VDR knockout mice, indicating that Vitamin D plays a significant role in negatively regulating the system [14,15,16,29]. While this study was carried out in the setting of proteinuric nephropathy and used levels of proteinuria as a statistical measure, similar concepts can be inferred in the setting of non-proteinuric DKD. This is because much of the pathophysiology is similar, and in some cases, non-proteinuric disease can be a precursor for the proteinuric form, as shown in several studies [14,15,16,29].
In a study that investigated the impact of RAAS inhibitors with and without vitamin D supplementation, it was found that Losartan in combination with paracalcitol prevented albuminuria and suppressed pro-fibrotic factors more effectively than either agent alone [https://pubmed.ncbi.nlm.nih.gov/18838678/]. This activation level was even more prominent in VDR knockout mice, indicating that Vitamin D plays a significant role in negatively regulating the system [14,15,16,29]. While this study was carried out in the setting of proteinuric nephropathy and used levels of proteinuria as a statistical measure, similar concepts can be inferred in the setting of non-proteinuric DKD. This is because much of the pathophysiology is similar, and in some cases, nonproteinuric disease can be a precursor for the proteinuric form, as shown in several studies [14,15,16,29]. More importantly, because some studies have suggested that the non-proteinuric form may be due to a more widespread inflammation [14,15] - rather than focusing on the podocytes of the glomerular filtration barrier - the direct reduction in levels of IL-6 and TNF-α by vitamin D would theoretically result in less inflammatory and ROS-caused damage in NPDKD [14]. The inflammatory response governed by these cytokines results in mitochondrial damage and subsequent ROS that cause damage to renal structures [12]. By binding to VDR, vitamin D induces anti-inflammatory cytokines and inhibits the secretion of pro-inflammatory ones [97]. Diabetics with renal damage exhibit downregulation of VDR, and therefore a decreased ability to counteract the pro-inflammatory actions of these cytokines [97]. By directly reducing the production of these cytokines, Vitamin D could play a critical role in reducing the overall inflammatory environment within the renal tubules and parenchyma, thereby reducing the risk of progression to more severe forms of renal disease [33]. Additionally, in a study of monocytes in diabetic patients, pre-treatment with vitamin D induced an increased expression of VDR mRNA [97]. This shows that treatment could negate, or mitigate, the negative effect of hyperglycemia on VDR expression. TGFβ is also directly implicated in the fibrosis that occurs in DKD via the stimulation of CTGF [47]. CTGF can be directly linked to the formation of fibrosis in studies of human biopsies. Its overexpression has been correlated to ECM accumulation in both the glomerulus and interstitium [47]. In mouse studies of diabetic nephropathy, blocking endogenous CTGF was found to have beneficial effects on the progression of renal damage [47]. By reducing CTGF’s stimulation by TGFβ, vitamin D could play a vital role in reducing the accumulation of ECM, and therefore fibrosis and progression of renal disease. An important impact of this inflammation is arterial stiffness and vasodilatory function, which can be measured by Pulse Wave Velocity (PWV) and Flow-Mediated Dilation (FMD), respectively [98]. Reduced FMD and increased PWV are seen commonly in patients with CKD, and multiple studies have identified a link to vitamin D status [same source as above].A clinical trial of 120 people with CKD 3-4 and vitamin D deficiency was carried out to compare these measures in patients randomized to high dose cholecaciferol or the placebo group [25]. They found a significant increase in FMD in the cholecalciferol group when compared to placebo [25].
The PENNY trail (Paricalcitol and Endothelial Function in Chronic Kidney Disease Trial) compared paricalcitol to placebo in patients with CKD 3-4 [99]. They similarly found a significant increase in FMD in the paricalcitol group. However, a trial out of Canada with patients randomized to placebo, calcifediol or calcitrol failed to show a significant reduction in PWV after baseline PWV values were balanced across groups [100]. These studies collectively indicate that there is a likely benefit of vitamin D on arterial stiffness, which has been associated with increased proteinuria, heart failure, and progression of CKD [98]. In the setting of NPDKD, this endothelial damage can occur early and eventually progress to the proteinuric form or lead to other complications. This data shows that there is significant value in using vitamin D to reduce the damage before the disease progresses to severe levels. While much of the above can also be of use in the proteinuric form, a key distinguishing feature of non-proteinuric disease could be the preponderance of macroangiopathic changes, as suggested by a 2023 study by Cherif et al. determined that vitamin D played an additive role with traditional risk factors in contributing to cardiovascular diseases [101]. As demonstrated in the Oh et al. study, Vitamin D sufficiency can reduce the risk of plaque formation by reducing the formation of foam cells and the uptake of cholesterol by macrophage s[94]. Again, we find mixed data on the actual clinical significance of this. In a meta-analysis carried out by Lu et al., that analyzed 17 RCTs and 21 observational studies on the effect of vitamin D therapy on mortality in adults with CKD stages 1-5, the observational studies showed a significant reduction in all-cause mortality that was consistent across subgroups of CKD stage, size of study, and route and type of Vitamin D agent used [102].
However, there was no significant difference found in the analysis of the RCTs [102]. Additionally, the Morrone trial in 2022, which investigated the impact of calcifediol in dialysis patients with comorbid hyperparathyroidism and vitamin D deficiency, found no significant reduction in major adverse cardiovascular events or cardiovascular death [102]. It is, however, important to note that these studies investigated patients with much more severe disease. The Opera trial in 2014, which investigated whether oral vitamin D reduced left ventricular mass, improved systolic and diastolic function, reduced the number of cardiovascular-related hospitalizations, and other secondary outcomes in patients with CKD 3-5. They found that, compared to placebo, paricalcitol treatment significantly reduced the number of cardiovascularrelated hospitalizations [103]. This indicates a potential benefit in earlier stages of CKD. While none of these studies directly investigated DKD, it is reasonable to infer that many of the same pathologic principles would apply. Although there is minimal research on the subject of Vitamin D Receptor Activators (VDRAs), specifically in the setting of NPDKD, they have been studied in CKD-MBD and are commonly used in its treatment. However, the results are mixed. There have been reports of cardio- and reno-protective effects in some studies [80], while others have failed to show benefits to cardiovascular function [104,105]. They also come with a significant risk of hypercalcemia and potentially CVC [106]. Additionally, a randomized clinical trial showed that cholecalciferol supplementation was protective of vascular outcomes, while other RCTs could not replicate this data. Importantly, for the treatment of CKD-MBD, 25-hydroxyvitamin D supplementation has been shown to suppress PTH and induce hypercalcemia less frequently than calcitriol. These drugs have also been studied in patients with CKD prior to the onset of CKDMBD In the two largest clinical trials that investigated fracture risk in CKD patients - the J-DAVID and PRIMO trials - there was a reduction in the raw number of fractures in the intervention groups, but neither reached statistical significance [98,104]. Multiple trials have shown an increase in Bone Mineral Density (BMD) with VDRAs and other forms of vitamin D among CKD patients. Post hoc analysis of the Vitamin D, Calcium, Lyon Study II (DECALYOS II), showed that women with an eGFR <45 mL/min that were taking cholecalciferol and calcium had a slower rate of BMD loss than those taking placebo. Additionally, smaller studies have shown that VDRAs increased BMD compared to placebo in patients with non-dialysis CKD. In total, this data shows that there is likely to be significant value of vitamin D supplementation to reduce adverse bone effects, such as osteopenia or fractures, of CKD and therefore DKD. However, the data is not strong enough to definitively determine that vitamin D supplementation would slow the progression of CKD-MBD. The promising results do suggest that further investigation is warranted. Based on the pathophysiology of this disease process, it is logical that Vitamin D could be a valuable component of a multi-drug regimen to slow its progression.
Discussion and Conclusion
In conclusion, vitamin D supplementation may offer a promising renoprotective strategy against the progression of non-proteinuric diabetic kidney disease, although further research is essential to establish its efficacy and optimize treatment regimens. The primary benefits are likely to be seen in slowing the progression of nephron injury in the early stages of the disease, reducing the progression to the proteinuric form as a result of the anti-inflammatory properties, its potential role in reducing cardiovascular morbidity, and promotion of bone health. Specifically, reduction in levels of IL-6 and TNF-a could contribute to a reduction in inflammatory damage associated with DKD. By reducing the overall levels of inflammation, you can in turn reduce the damage to mitochondria and subsequent reactive oxygen species production. This antiinflammatory action may be of particular importance in NPDKD in comparison to PDKD due to the theory that NPDKD is related to more widespread inflammation. However, given that much of the mitochondrial damage occurs as a result of hyperglycemia, glycemic control must be adequately addressed for any substantial impact to be seen. As studies have shown, there is a significant benefit to arterial compliance at healthy vitamin D levels. This is especially important for the control of hypertension, and reduction in cardiovascular morbidity and mortality. Vitamin D and its analogs can be used early in treatment to maintain vascular compliance.
Due to the increased association with macroangiopathic compared to microangiopathic issues in non-proteinuric disease, this mechanism could be crucial in uncovering the therapeutic benefit of Vitamin D supplementation. However, because kidney disease itself is primarily a microangiopathic issue, this benefit would be indirectly related to the kidney impairment itself. This underscores the importance of using Vitamin D therapy only as an adjunct to current treatment regimens and not as a standalone therapy. The importance of early initiation of therapy is seen in the studies involving dialysis patients where no significant reduction in cardiac mortality was seen. This suggests that there may be a tipping point where the benefits are not significant enough to offset the damage that has already been done by the disease process. Similarly, in the setting of bone disease, the most conclusive benefits appear to be related to the maintenance or improvement of BMD, rather than the treatment of CKD-MBD. This benefit is two-fold - maintenance of PTH homeostasis and directly in bone mineralization. The data on VDRAs in the setting of CKD-MBD show significant risk of hypercalcemia and other complications, while not significantly improving outcomes. This further underscores that any treatment with vitamin D or its analogs should be initiated early in the disease process. Concurrently, established therapies such as ACE inhibitors, ARBs, and SGLT2 inhibitors continue to play a critical role in managing diabetic kidney disease and should not be ignored in favor of vitamin D supplementation as there is insufficient evidence to suggest vitamin D can operate efficiently as a standalone therapy. A multifactorial approach that incorporates vitamin D supplementation alongside these traditional therapies may ultimately enhance patient outcomes.
Gaps in Research and Future Directions (last paragraph after discussion)
Despite the potential benefits of vitamin D, significant gaps remain in the current literature, particularly regarding the optimal dosage and long-term effects of supplementation in non-proteinuric DKD. Future research should focus on randomized controlled trials that assess the effectiveness of vitamin D in diverse populations with varying stages of kidney disease [45]. Furthermore, investigating the synergistic effects of vitamin D alongside established renoprotective agents like ACEis, ARBs, and SGLT2 inhibitors may provide new insights into comprehensive management strategies for non-proteinuric DKD.
References
- Stevens PE, Ahmed SB, Carrero JJ, KDIGO (2024) Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 105: S117-S314.
- (2023) Writing Group for the CKD Prognosis Consortium. Estimated Glomerular Filtration Rate, Albuminuria, and Adverse Outcomes: An Individual-Participant Data Meta-Analysis. JAMA 330: 1266-1277.
- Webster AC, Nagler EV, Morton RL, Masson P (2017) Chronic Kidney Disease. Lancet Lond Engl 389: 1238-1252.
- Gupta S, Dominguez M, Golestaneh L (2023) Diabetic Kidney Disease: An Update. Med Clin North Am107: 689-705.
- Selby NM, Taal MW (2020) An updated overview of diabetic nephropathy: Diagnosis, prognosis, treatment goals and latest guidelines. Diabetes Obes Metab. 1: 3-15.
- Barreto DV, Barreto FC, Liabeuf S (2010) Plasma interleukin-6 is independently associated with mortality in both hemodialysis and predialysis patients with chronic kidney disease. Kidney Int. 77: 550-556.
- Zhang L, Xu F, Hou L (2024) IL-6 and diabetic kidney disease Front Immunol.19: 15
- Meng XM, Nikolic-Paterson DJ, Lan HY (2014) Inflammatory processes in renal fibrosis. Nat Rev Nephrol 10: 493-503.
- Black LM, Lever JM, Agarwal A (2019) Renal Inflammation and Fibrosis: A Double-edged Sword. J Histochem Cytochem67: 663-681.
- Sánchez-López E, Rayego S, Rodrigues-Díez R (2009) CTGF Promotes Inflammatory Cell Infiltration of the Renal Interstitium by Activating NF-κB. J Am Soc Nephrol JASN 20:1513-1526.
- Forbes JM, Thorburn DR (2018) Mitochondrial dysfunction in diabetic kidney disease. Nat Rev Nephrol 14: 291-312.
- X Hu, W Liu,Y Yan (2019 )Vitamin D protects against diabetic nephropathy: Evidence-based effectiveness and mechanism. Eur J Pharmacol 845: 91-98.
- Yamanouchi M, Furuichi K, Hoshino J (2019) Nonproteinuric Versus Proteinuric Phenotypes in Diabetic Kidney Disease: A Propensity Score-Matched Analysis of a Nationwide, Biopsy-Based Cohort Study. Diabetes Care42: 891-902.
- Kopel J, Pena-Hernandez C, Nugent K (2019) Evolving spectrum of diabetic nephropathy. World J Diabetes 10: 269-279.
- Yamanouchi M, Furuichi K, Hoshino J, Ubara Y, Wada T (2020) et al. Nonproteinuric diabetic kidney disease. Clin Exp Nephrol 24: 573-581.
- Pal R, Bhadada SK (2023) Ages accumulation with vascular complications, glycemic control and metabolic syndrome: A narrative review. Bone, 176.
- Galuška D, Pácal L, Kaňková K (2021) Pathophysiological Implication of Vitamin D in Diabetic Kidney Disease. Kidney Blood Press Res 46: 152-161.
- Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD (1997) et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 99: 342-348
- Zhang Z, Sun L, Wang Y, Ning G, Minto AW (2008) et al. Renoprotective role of the vitamin D receptor in diabetic nephropathy. Kidney Int 73: 163-171.
- Lin YC, Chang YH, Yang SY, Wu KD, Chu TS (2018) et al. Update of pathophysiology and management of diabetic kidney disease. Journal of the Formosan Medical Association 117: 662-675.
- Hsu FY, Lin FJ, Ou HT, Huang SH, Wang CC (2017) et al. Renoprotective Effect of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers in Diabetic Patients with Proteinuria. Kidney Blood Press Res 42: 358-368.
- Yang Y, Lei Y, Liang Y, Fu S, Yang C, (2021) et al.Vitamin D protects glomerular mesangial cells from high glucose-induced injury by repressing JAK/STAT signaling. Int Urol Nephrol. 53: 1247-1254.
- Delrue C, Speeckaert R, Delanghe JR, Prytuła A, Speeckaert MM (2024) et al. Investigating Vitamin D-Binding Protein’s Role in Childhood Health and Development. International Journal of Molecular Sciences 25: 6272.
- Hesp AC (2020) The role of renal hypoxia in the pathogenesis of diabetic kidney disease: a promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int 98: 579-589 Klag.
- Jamerson K (1996) The effect of blood pressure reduction on end stage renal disease. Am J Hypertens 9: 60s-64s.
- Penno G, Solini A, Bonora E (2018) Defining the contribution of chronic kidney disease to all-cause mortality in patients with type 2 diabetes: the Renal Insufficiency and Cardiovascular Events (RIACE) Italian Multicenter Study. Acta Diabetol 55: 603-612.
- Thomas MC, Macisaac RJ, Jerums G, Weekes A, Moran J (2009) et al. Nonalbuminuric renal impairment in type 2 diabetic patients and in the general population (national evaluation of the frequency of renal impairment cO-existing with NIDDM [NEFRON] 11). Diabetes Care 32: 1497-1502.
- Dwyer JP, Parving HH, Hunsicker LG, Ravid M, Remuzzi G (2012) et al. Renal Dysfunction in the Presence of Normoalbuminuria in Type 2 Diabetes: Results from the DEMAND Study. Cardiorenal Med 2: 1-10.
- Oshima M, Shimizu M, Yamanouchi M (2021) Trajectories of kidney function in diabetes: a clinicopathological update. Nat Rev Nephrol 17: 740-750.
- Galichon P, Lannoy M, Li L, Serre J, Vandermeersch S (2024) et al. Energy depletion by cell proliferation sensitizes the kidney epithelial cells to injury. Am J Physiol Renal Physiol 326: F326-F337.
- Barutta F, Bellini S, Gruden G (2022) Mechanisms of podocyte injury and implications for diabetic nephropathy. Clin Sci (Lond) 136: 493520.
- Huang HY, Lin TW, Hong ZX, Lim LM (2023) Vitamin D and Diabetic Kidney Disease. Int J Mol Sci 24: 3751.
- Robles NR, Villa J, Gallego RH (2015) Non-Proteinuric Diabetic Nephropathy. J Clin Med 4:1761-1773.
- Shimizu M, Furuichi K, Yokoyama H (2014) et al. “Kidney lesions in diabetic patients with normoalbuminuric renal insufficiency,” Clinical and Experimental Nephrology 18: pp 305-312.
- Ekinci EI, Jerums G, Skene A (2013) “Renal structure in normoalbuminuric and albuminuric patients with type 2 diabetes and impaired renal function,” Diabetes Care 36: 3620-3626.
- Zoccali C, Mallamaci F (2019) “Nonproteinuric progressive diabetic kidney disease,” Current Opinion in Nephrology and Hypertension 28: 227-232.
- Donate-Correa J, Luis-Rodríguez D, Martín-Núñez E 2020 “Inflammatory targets in diabetic nephropathy,” Journal of Clinical Medicine 9: 458.
- Chawla V, Roshan B (2014) “Non-proteinuric diabetic nephropathy,” Current Diabetes Reports 14: 529.
- MacIsaac RJ, Tsalamandris C, Panagiotopoulos S, Smith TJ, McNeil KJ (2004) et al. “Nonalbuminuric renal insufficiency in type 2 diabetes,” Diabetes Care 27: 195-200.
- Silva R, Meng C,Coentrão L (2017) “Diabetic nephropathy and its two phenotypes: the proteinuric and non-proteinuric,” Portuguese Journal of Nephrology & Hypertension 31: 122-131.
- Thakar CV, Christianson A, Himmelfarb J, Leonard AC (2011) “Acute kidney injury episodes and chronic kidney BioMed Research International 13 disease risk in diabetes mellitus,” Clinical Journal of the American Society of Nephrology 6: 2567–2572.
- Sykes L, Asar O, Ritchie J (2019) “The influence of multiple episodes of acute kidney injury on survival and progression to end stage kidney disease in patients with chronic kidney disease,” PLoS One14: e0219828,
- Sánchez-Lozada LG, Lanaspa MA, Cristóbal-García M (2013) “Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations,” Nephron Experimental Nephrology 121: e71-e78.
- Rabadi MM, Kuo MI, Ghaly CT (2011) “Interaction between uric acid and HMGB1 translocation and release from endothelial cells,” American Journal of Physiology Renal Physiology 302: F730–F741.
- Kang DH, Park SK, Lee IK, Johnson RJ (2005) “Uric acidinduced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells,” Journal of the American Society of Nephrology 16: 3553-3562.
- Corry DB, Eslami P, Yamamoto K, Nyby MD, Makino v (2008) et al.“Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system,” Journal of Hypertension26:269-275.
- Zhou Y, Fang L, Jiang L (2012) “Uric acid induces renal inflammation via activating tubular NF-κB signaling pathway,” PLoS One 7: e39738,.
- Ryu S, Kim M. J, Shin H. S(2013) “Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease,” American Journal of PhysiologyRenal Physiology 304: F471-F480,
- Niewczas MA, Ficociello LH, Johnson AC (2009) “Serum concentrations of markers of TNFalpha and Fas-mediated pathways and renal function in nonproteinuric patients with type 1 diabetes,” Clinical Journal of the American Society of Nephrology 4: 62–70,
- Akcay A, Nguyen Q,Edelstein C. L (2009) “Mediators of inflammation in acute kidney injury,” Mediators of Inflammation2009: 137072,.
- Vielhauer V, Mayadas TN (2007)“Functions of TNF and its receptors in renal disease: distinct roles in inflammatory tissue injury and immune regulation,” Seminars in Nephrology 27: 286-308.
- Ernandez T, Mayadas TN (2009) “Immunoregulatory role of TN alpha in inflammatory kidney diseases,” Kidney International 76 :276-276.
- Buyadaa O, Magliano DJ, Salim AN, Koye D,Shaw JE (2020) “Risk of rapid kidney function decline, all-cause mortality, and major cardiovascular events in nonalbuminuric chronic kidney disease in type 2 diabetes,” Diabetes Care43:122-129.
- Fiorini F, Barozzi L (2007) “The role of ultrasonography in the study of medical nephropathy,” Journal of Ultrasound 10:161-167.
- Abd el Dayem S, el Bohy AE, el Shehaby A (2017)“Value of the intrarenal arterial resistivity indices and different renal biomarkers for early identification of diabetic nephropathy in type 1 diabetic patients,” Journal of Pediatric Endocrinology and Metabolism 5:188-192.
- Spatola L, Andrulli S (2016) “Doppler ultrasound in kidney diseases: a key parameter in clinical long-term follow-up,” Journal of Ultrasound 19: 243-250.
- Mancini M, Masulli M, Liuzzi R (2013) “Renal duplex sonographic evaluation of type 2 diabetic patients,” Journal of Ultrasound in Medicine 32: 1033-1040.
- Christakos S, Ajibade DV, Dhawan P, Fechner AJ, Mady LJ (2010) Vitamin D: metabolism. Endocrinol Metab Clin North Am 39: 243-253, table of contents.
- Bikle DD (2014) Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol 21: 319-329.
- Breslau NA (1988) Normal and abnormal regulation of 1,25-(OH)2D synthesis. Am J Med Sci 296: 417-425.
- Khundmiri SJ, Murray RD, Lederer E (2016) PTH and Vitamin D. Compr Physiol6: 561-601.
- de Oliveira E Silva Ullmann T, Ramalho BJ, Laurindo LF (2023) Effects of Vitamin D Supplementation in Diabetic Kidney Disease: A Systematic Review. J Ren Nutr Off J Counc Ren Nutr Natl Kidney Found 33: 618-628.
- Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G (2016) et al.Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol Rev 96: 365-408.
- Agoro R, White KE (2023) Regulation of FGF23 production and phosphate metabolism by bone-kidney interactions. Nat Rev Nephrol 19: 185-193
- Sugimoto T, Ritter C, Morrissey J, Hayes C, Slatopolsky E (1990) Effects of high concentrations of glucose on PTH secretion in parathyroid cells. Kidney Int. 37:1522-1527.
- Jankowski M, Biber J, Murer H (1999) PTH-induced internalization of a type IIa Na/Pi cotransporter in OK-cells. Pflugers Arch438: 689-693.
- Yamada S, Nakano T ( 2023) Role of Chronic Kidney Disease (CKD)-Mineral and Bone Disorder (MBD) in the Pathogenesis of Cardiovascular Disease in CKD. J Atheroscler Thromb 30: 835-850.
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, (2006) et al Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444: 770-774.
- Saar-Kovrov V, Donners MMPC (2021) van der Vorst EPC. Shedding of Klotho: Functional Implications in Chronic Kidney Disease and Associated Vascular Disease. Front Cardiovasc Med, 7: 617842.
- Oliveira RB, Cancela AL, Graciolli FG, Dos Reis LM, Draibe SA (2010) Early control of PTH and FGF23 in normophosphatemic CKD patients: a new target in CKD-MBD therapy? Clin J Am Soc Nephrol, 5: 286291.
- Drüeke TB.2021 Oct 18. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E (2000) et al. Hyperparathyroidism in Chronic Kidney Disease. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.;2000
- Shah A, Hashmi MF, Aeddula NR (2025) Chronic Kidney DiseaseMineral Bone Disorder (CKD-MBD). In: StatPearls. StatPearls Publishing; 2025.
- Hu L, Napoletano A, Provenzano M, (2022) Mineral Bone Disorders in Kidney Disease Patients: The Ever-Current Topic. Int J Mol Sci 23: 12223.
- Dharmarajan S, Speer MY, Pierce K, Lally J, Leaf EM (2021) et al. Role of Runx2 in Calcific Aortic Valve Disease in Mouse Models. Front Cardiovasc Med8: 687210.
- Son BK, Kozaki K, Iijima K, Eto M, Kojima T, Ota H (2006) et al.Statins protect human aortic smooth muscle cells from inorganic phosphateinduced calcification by restoring Gas6-Axl survival pathway. Circ Res 98: 1024-1031.
- Liu WC, Wu CC, Hung YM, Liao MT, Shyu JF (2016 ) et al. Pleiotropic effects of vitamin D in chronic kidney disease. Clin Chim Acta 453: 1-12.
- Ketteler M, Block GA, Evenepoel P (2017) Executive summary of the 2017 KDIGO Chronic Kidney Disease–Mineral and Bone Disorder (CKD-MBD) Guideline Update: what’s changed and why it matters. Kidney Int 92: 26-36.
- Zac-Varghese S, Winocour P (2018) Managing diabetic kidney disease. Br Med Bull. 125: 55-66.
- Stathi D, Fountoulakis N, Panagiotou A (2023) Impact of treatment with active vitamin D calcitriol on bone turnover markers in people with type 2 diabetes and stage 3 chronic kidney disease. Bone 166:116581.
- Joergensen C, Gall MA, Schmedes A, Tarnow L, Parving HH (2010) et al. Vitamin D Levels and Mortality in Type 2 Diabetes. Diabetes Care 33: 2238-2243.
- Chertow BS, Sivitz WI, Baranetsky NG, Clark SA, Waite A (1983) et al. Cellular mechanisms of insulin release: the effects ofvitamin D deficiency and repletion on rat insulin secretion. Endocrinol-ogy 113: 1511-1518.
- Christakos S, Friedlander EJ, Frandsen BR, Norman AW (1979) Studies on the mode of action of calciferol. XIII. Development of aradioimmunoassay for vitamin D-dependent chick intestinal calciumbinding protein and tissue distribution. Endocrinology 104: 1495-1503.
- Allegra V, Mengozzi G, Martimbianco L, Vasile A (1990) Glucoseinduced insulin secretion in uremia: effects of aminophylline infusionand glucose loads. Kidney Int 38: 1146 -1150.
- Alvestrand A, Mujagic M, Wajngot A, Efendic (1989) S Glucoseintolerance in uremic patients: the relative contributions of impaired-cell function and insulin resistance. Clin Nephrol 31: 175– 183.
- Lowrie EG, Soeldner JS, Hampers CL, Merrill JP (1970) Glucosemetabolism and insulin secretion in uremic, prediabetic, and normalsubjects. J Lab Clin Med 76: 603- 615.
- Allegra V, Luisetto G, Mengozzi G, Martimbianco L, Vasile A (1994) et al.Glucose-induced insulin secretion in uremia: role of 1,25(HO)2 -vitamin D3. Nephron 68: 41- 47.
- Quesada JM, Martin-Malo A, Santiago J, Hervas F, Martinez ME (1990) et al. Effect of calcitriol on insulinsecretion in uraemia. Nephrol Dial Transplant 5: 1013-1017.
- Schwartz GG, Eads D, Rao A, Cramer SD, Willingham MC (2004) et al. Pancreatic cancer cells express 25-hydroxyvitamin D-1-hydrox- ylase and their proliferation is inhibited by the prohormone 25-hydroxyvitamin D3 . Carcinogenesis 25: 1015-1026.
- Kim J, Nam JS, Kim H, Lee HS, Lee JE (2021) No effect of vitamin D supplementation on metabolic parameters but on lipids in patients with type 2 diabetes and chronic kidney disease. Int J Vitam Nutr Res Int Z Vitam- Ernahrungsforschung J Int Vitaminol Nutr 91: 649-658.
- Oh J, Weng S, Felton SK (2009) 1,25(OH)2Vitamin D inhibits foam cell formation and suppresses macrophage cholesterol uptake in patients with type 2 diabetes. Circulation.;120: 687-698.
- Tian B, Deng Y, Cai Y, Han M, Xu G (2022) Efficacy and safety of combination therapy with sodium-glucose cotransporter 2 inhibitors and renin-angiotensin system blockers in patients with type 2 diabetes: a systematic review and meta-analysis. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc 37: 720-729.
- Cai Y, Shi W, Xu G (2020) The efficacy and safety of SGLT2 inhibitors combined with ACEI/ARBs in the treatment of type 2 diabetes mellitus: A meta-analysis of randomized controlled studies. Expert Opin Drug Saf 19: 1497-1504.
- Yang M, Shen Z, Chen D, Gan H, Shen Q (2012)Effects of 1,25(OH)(2)D (3) on the expressions of vitamin D receptor, STAT5 and cytoskeletal rearrangement in human monocytes incubated with sera from type 2 diabetes patients and diabetic nephropathy patients with uremia. Inflamm Res 61: 511-520.
- Yeung WG, Toussaint ND, Badve SV (2024) Vitamin D therapy in chronic kidney disease: a critical appraisal of clinical trial evidence. Clin Kidney J 17:sfae227.
- Zoccali C, Curatola G, Panuccio V, Tripepi R, Pizzini P (2014) Paricalcitol and endothelial function in chronic kidney disease trial. Hypertension. 64: 1005-1011.
- J-DAVID Investigators; Shoji T, Inaba M, Fukagawa M, Ando R, Emoto M (2018) et al. Effect of Oral Alfacalcidol on Clinical Outcomes in Patients Without Secondary Hyperparathyroidism Receiving Maintenance Hemodialysis: The J-DAVID Randomized Clinical Trial. JAMA 320: 2325-2334.
- Cherif AB, Bennouar S (2023) PREDICTIVE FACTORS OF MACROANGIOPATHY IN HYPERTENSIVE DIABETIC PATIENTS WITH VITAMIN D DEFICIENCY. J Hypertens. 41: e200.
- Morrone L, Palmer SC, Saglimbene VM, Perna A, Cianciolo G Strippoli (2022) GFM; Mineral Metabolism Study Group of the Italian Society of Nephrology. Calcifediol supplementation in adults on hemodialysis: a randomized controlled trial. J Nephrol 35: 517-525.
- Wang AY, Fang F, Chan J, Wen YY, Qing S (2014) Effect of paricalcitol on left ventricular mass and function in CKD--the OPERA trial. J Am Soc Nephrol. 25: 175-186.
- Thadhani R, Appelbaum E, Pritchett Y, Chang Y, Wenger J (2012) et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: the PRIMO randomized controlled trial. JAMA 307: 674-684.
- Wang AY, Fang F, Chan J, Wen YY, Qing S (2014) Effect of paricalcitol on left ventricular mass and function in CKD--the OPERA trial. J Am Soc Nephrol25: 175-186.
- Cozzolino M, Bernard L, Csomor PA (2021) Active vitamin D increases the risk of hypercalcaemia in non-dialysis chronic kidney disease patients with secondary hyperparathyroidism: a systematic review and meta-analysis. Clin Kidney J 14: 2437-2443.
- Bishop CW, Strugnell SA, Csomor P, Kaiser E, Ashfaq A (2022) Extended-Release Calcifediol Effectively Raises Serum Total 25-Hydroxyvitamin D Even in Overweight Non dialysis Chronic Kidney Disease Patients with Secondary Hyperparathyroidism. Am J Nephrol53: 446-454.
- Bosworth C, de Boer IH, Targher G, Kendrick J, Smits G, (2011) et al. The effect of combined calcium and cholecalciferol supplementation on bone mineral density in elderly women with moderate chronic kidney disease. Clin Nephrol 77: 358-365.
- Przedlacki J, Manelius J, Huttunen K (1995) Bone mineral density evaluated by dual-energy X-ray absorptiometry after one-year treatment with calcitriol started in the predialysis phase of chronic renal failure. Nephron 69: 433-437.
- Rix M, Eskildsen P, Olgaard K (2004) Effect of 18 months of treatment with alfacalcidol on bone in patients with mild to moderate chronic renal failure. Nephrol Dial Transplant 19: 870-876.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.