The Whole Exome Sequencing (WES) Data Identify the Common Genetic Alterations in Stomach and Colorectal Cancers as Dual Primary Cancers
by Rabindra Bera 1, Sangita Dan1, Soma Sett1, Sparsha Dey1, Palash Dhara1, Krishn Prasad Sahoo1, Parvez Alam1, Jayanta Chakrabarti2,
Subhranshu Mandal3, Sankar Sengupta3, Chandan Mandal 1*
1Department of Molecular Biology, Chittaranjan National Cancer Institute, India
2Department of Surgical Oncology, Chittaranjan National Cancer Institute, India
3Department of Laboratory Services, Chittaranjan National Cancer Institute, India
*Corresponding author: Chandan Mandal, Department of Molecular Biology, Chittaranjan National Cancer Institute, Kolkata, West Bengal, India
Received Date: 04 March, 2026
Accepted Date: 13 March, 2026
Published Date: 24 March, 2026
Citation: Bera R, Dan S, Sett S, Dey S, Dhara P, et al. (2026) The Whole Exome Sequencing (WES) data identify the common genetic alterations in Stomach and colorectal cancers as dual primary Cancers. Ann med clin Oncol 9: 171. DOI: https://doi.org/10.29011/2833-3497.000171
Abstract
Stomach Cancer (SC) and Colorectal Cancer (CRC) often coexist as Dual Primary Cancers (DPCs), significantly impacting global oncology. In 2020, gastrointestinal cancers affected nearly 5 million people, causing 3.5 million deaths. Among these, stomach and colorectal cancers are most prevalent, especially with advancing age. Understanding synchronous and metachronous DPCs is crucial for improved treatment and survival. Whole Exome Sequencing (WES) aids in identifying germline mutations and molecular mechanisms underlying these cancers. In this study, a DPC patient’s DNA was analysed using Next Generation Sequencing (NGS), examining 437 genes. Thirty-one genes, including exon and intron variants, showed mutations. Notably, the MLH1 gene emerged as a key biomarker with a 50% variant allele frequency and a pathogenic stop-gained mutation (NM_000249.4: c.2141G>A) confirmed by ClinVar. Immunohistochemistry and clinical analyses of MLH1 and associated genes provided insights into molecular pathways, treatment duration, and patient outcomes in SC and CRC in DPCs.
Keywords: Stomach Cancer (SC), Colorectal Cancer (CRC), Whole Exome Sequencing (WES), Mis-match Repair (MMR), RAS/MAPK Pathways, MLH1 Genes, Immunohistochemistry (IHC).
Introduction
The Co-existence of Stomach Cancers (SC) and Colorectal Cancers (CRC) as Dual Primary cancers (DPCs) presents a major challenge in oncology. The Incidence of CRC in patients with SC is approximately 6.4%, while SC occurs in 4.4% of CRC cases [1]. CRC ranks 11.4% among rapidly developing multiple primary tumors, whereas SC ranges from 1-4% as a secondary cancer. Most SC cases are Sporadic, but familial aggregation also contributes, showing both autosomal dominant and autosomal recessive inheritance patterns. DPCs are classified as synchronous and metachronous: the former arises from environmental and genetic factors, while the latter develops from long-term carcinogenic or treatment-related effects [2]. Germline mutations in tumor suppressor and DNA repair genes play a key role in DPC pathogenesis.
Next-generation sequencing (NGS) and Whole-Exome Sequencing (WES) have revealed mutations in the DNA mismatch repair (dMMR) genes, such as MLH1, MSH2, MSH6, and PMS2, as well as other DNA repair genes like EXO1 and ERCC2, which are crucial in developing DPC [3]. Notably, the MLH1 mutation on chromosome 3 is linked to Lynch Syndrome. Mutation in Signalling Pathways- RAS/MAPK Pathways, apoptotic, Cytoskeleton, and Immune regulatory pathways also contribute the tumor progression [4].
This study integrates genomic and clinical data from DPC patients to identify the mutations, copy number variations and allele frequency changes in both SC and CRC. These findings highlight the key molecular mechanisms, reveal the potential biomarkers for early detection and propose targeted therapeutic strategies aimed at improving treatment outcomes and patient survival in dual primary cancers [5].
Clinical Presentation and Pathological Data
We report a case of a 55-year-old man, who was admitted to Chittaranjan National Cancer Hospital, West Bengal, India, who was a smoker and brought typhoid, who was evaluated for epigastric pain and weakness for 6-7 months, associated with anorexia, weight loss, and occasional Constipation. Endoscopy revealed gastric ulcers at the posterior of the corpus, whose biopsy report came out to be poorly or moderately differentiated adenocarcinoma. Positron Emission Tomography-Computed Tomography (PET-CT) scan reveals that lesions in the body, stomach, and 1/3rd of the descending colon are affected with synchronous carcinoma, lobulated enhancing lesions in the anal canal, and the mid part of the descending colon.
A colonoscopy revealed a malignant-looking stricture in the ascending colon, with a biopsy report indicating poorly differentiated adenocarcinoma. Intraoperative findings (Figure 1) showed a gastric ulcer approximately 4 cm in size on the posterior wall along the greater curvature. The gastric biopsy (67/23) was examined in fragments, revealing gastric mucosa with ulceration characterised by dense acute inflammatory cells and fibrin; the submucosa showed dense lymphoplasmacytic infiltrate. The colonic biopsy (68/23) (Figure 1) consisted of multiple fragments from the colonic Tirone section, which showed mucosa with superficial ulceration. There was a dense inflammatory infiltrate in the submucosa, along with crypt abscesses and focal cryptitis. The coloscopy report indicated that the sigmoid colon had a circumferential, friable, proliferative lesion causing complete occlusion of the lumen.
|
Pathological Data of DPC Pati |
ent |
|
|
Biochemistry |
Haemoglobin |
5.1gm/dl |
|
Creatinine |
0.9 mg/dl |
|
|
Sodium |
136 mmol/L |
|
|
Potassium |
4.1 mmol/L |
|
|
Chloride |
101 mmol/L |
|
|
Bilirubin |
0.3 mg/dl |
|
|
Protein |
5.5 g/dl |
|
|
Calcium |
8.1 mg/L |
|
|
Phosphorus |
4.8 mg/L |
|
|
Serum Magnesium |
2 mg/L |
|
|
Serum glutamic–oxaloacetic transaminase (SGOT) |
17 u/L |
|
|
Serum glutamic–pyruvic transaminase (SGPT) |
16 u/l |
|
|
Haematology |
C-Reactive protein (CRP) |
23.4 mg/L |
|
Neutrophil Lymphocyte Ratio (NLR) |
4.4 |
|
|
Monocyte Lymphocyte Ratio (MLR) |
0.25 |
|
|
Platelet Lymphocyte Ratio (PLR) |
0.29 |
|
|
Serology |
Non-R |
eactive |
|
Cell Culture and Growth |
No G |
rowth |
Table 1: Pathological Data of DPC Patient.
Immunohistochemistry (IHC) and Endoscopy Profile
In a histopathological study, we show a large biopsy of this patient, received specimen labelled as ‘distal gastrectomy’ measuring (16 x 7 x 3) Cm. Specimen measures 20 cm along the greater curvature and 7 cm along the lesser curvature. Greater omentum measuring (38 x 15 x 0.5) and lesser omentum measuring (12 x 6 x 0.4). whereas, a small biopsy taken from the left colonic proliferative lesion, received specimen labelled as “Biopsy taken from left colonic proliferative lesion” comprising of multiple greyish white tissue pieces, altogether measuring (0.5 x 0.3 x 0.1) cm. In Immunohistochemistry data of both gastric and colorectal Cancer, we measure the pathogenic genes, like MLH1, that have greater malignancy in these DPCs. Therefore, MLH1 genes are selected as a biomarker in stomach and colorectal carcinoma, and it’s reported as a pathogenic variant.
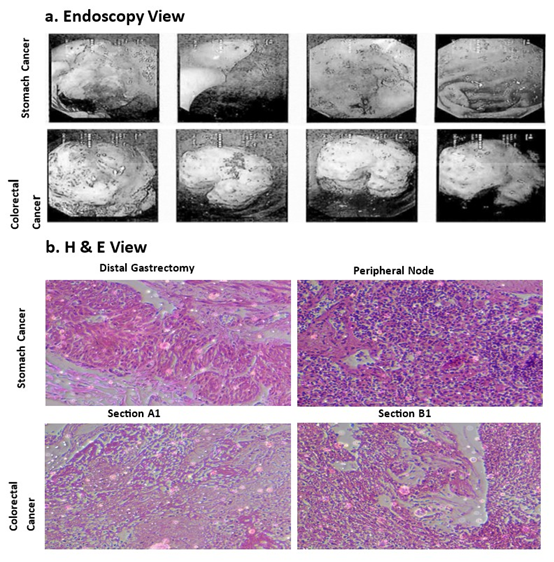
Figure 1: (a). Endoscopy picture of patient Stomach (SCs) and Colorectal cancer (CRCs). (b). H & E picture of distal gastrectomy and peripheral node expression in these DPCs.
Methodology
Sample Preparation
DNA is extracted using commercial blood extraction kits (Qiagen) from peripheral venous whole blood obtained in an EDTA tube or other body fluids for samples. The DNA thus obtained underwent multiple quality assessments. The in-house validation ensured that the samples passed all established laboratory QC metrics. This includes sample QC and library QC. Samples that pass the QC are taken further for library preparation.
Library preparation and Assay
Sequencing libraries were prepared using enzymatic shearing and ligation-based library preparation, following the library preparation protocols provided by Integrated DNA Technologies (xGenTM DNA Library prep EZ). Subsequently, the libraries were subjected to capture and multiplexing processes by the IDT capture protocol (xGen Hyb Panel v2). After the quality check, the enriched libraries were then subjected to sequencing on an Illumina Novaseq instrument, using 2 x 150 base, paired-end reads for analysis.
Quality Assessment
The quality of nucleic acids is assessed using Fluorometry (Thermo Scientific Qubit) and Automated gel electrophoresis (Agilent Tape Station). Samples after extraction, post-fragmentation, post-index PCR, and final amplified libraries after capture are assessed for QC. Samples that pass the minimum QC are only taken for the next step of the process.
Variant Calling and Annotation
Alignment, post-processing, and default quality filtered variant calling were executed on the Illumina DRAGEN Bio-IT platform developed by Illumina Inc., USA. The analysis was facilitated by the dynamic read Analysis for Genomics (DRAGEN) v4.2.4 pipeline using the GRCh38 (hg38) human reference genome. The resulting variants were further annotated using Varseq, and annotation sources included the human genome assembly GRCh38.p14, Refseq v110, regulatory build 1.0, polyphen v2.2.3, SIFT v6.2.1, dbSNP v154, Clinvar v2022-09, 1000 genomes phase3, gnomAD exomes r2.1.1, gnomAD genomes r3.1.2 and dbNSFP v4.4.c. These databases collectively contribute to a comprehensive and accurate interpretation of the genetic variants, thereby enhancing the clinical significance of the reported results.
Variant Assessment
Annotated variants were assessed and filtered to retain highconfidence variants based on the following criteria: minimum Read Depth (DP) of 10x, Genotype Quality (GQ) greater than 20, and minor allele frequency less than 1% in the known germline population databases. Further, Variants reported to be benign in ClinVar were filtered out, followed by ACMG classification to retain the pathogenic, likely pathogenic, and VUS candidates only. The variant interpretation was performed using the ACMG variant classification guidelines. The following recessive gene thresholds were used: Common Allele Frequency: 0.01, High for Disorder Allele Frequency: 0.0015; Extremely rare allele frequency: 0.0002; The following dominant gene threshold were used: Common Allele frequency: 0.005 High for Disorder Allele Frequency: 0.0005; Extremely Rare Allele Frequency: 0.0001; sub-populations were excluded from consideration if the total allele number failed to exceed 2000.
Results
Total Mutated genes Revealed by Whole Exome Sequencing (WES) data in these DPCs
The germline testing found that a total of 31 genes had altered sequences among the genes analysed in DPCs. In 31 genes, almost 14 genes are exon-specific genes, and 17 are intron-specific genes.
In 14 exon-specific genes, only one is a relevant biomarker (MLH1), which is characterised as a pathogenic variant (p. Trp714Ter; VAF=50.00 %) by germline analysis; the other 13 exon-specific genes are not relevant for genomic significance, so they are characterised as Non-pathogenic.
|
Gene/ Transcript |
Variant |
Zygosity |
Coverage/VAF |
Inheritance |
Variant Classification |
|
NM_000249.4 |
c.2141G>A p.Trp714Thr 3:37050523 rs63751022 Exon 19 |
Heterozygous |
28/50% |
Autosomal Dominant |
Pathogenic |
|
SPTA1 NM_003126.4 |
c.4031C>A p.Thr1344Asn 1:158645351 Exon 29 |
Heterozygous |
45/44.44% |
Autosomal Heterozygous |
Non-Pathogenic |
|
DIS3L2 NM_152383.5 |
c.393+53C>T p. Pro88Leu 2:232024329 rs202059499 Exon 4 |
Heterozygous |
25/44.00% |
Autosomal Recessive |
Non-Pathogenic |
|
NBEAL2 NM_015175.3 |
c.6865C>T p. Arg2289Trp 3:47006009 Rs956571690 Exon 43 |
Heterozygous |
38/52.63% |
Autosomal Recessive |
Non-Pathogenic |
|
NBEAL2 NM_015175.3 |
c.8201C>T p. Ser2734Phe 3:47009256 Rs768122212 Exon 54 |
Heterozygous |
33/69.7% |
Autosomal Recessive |
Non-Pathogenic |
|
PDGFRA NM_006206.6 |
c.1099G>A p. Val367Met 4:54267719 Rs147982027 Exon 7 |
Heterozygous |
37/35.14% |
Autosomal Heterozygous |
Non-Pathogenic |
|
SBDS NM_016038.4 |
C.437C>T p. Thr146Ile 7:66993239 Exon 3 |
Heterozygous |
32/37.5% |
Autosomal Recessive |
Non-Pathogenic |
|
CDKN2A NM_000077.5 |
c.232C>T p. Leu78Phe 9:21971127 Rs1563889606 Exon 2 |
Heterozygous |
52/46.15% |
Autosomal Dominant |
Non-Pathogenic |
|
ANKRD26 NM_014915.3 |
c.3447A>G p. Gln1149= 10:27035003 Rs1426031873 |
Heterozygous |
55/49.09% |
Autosomal Dominant |
Non-pathogenic |
|
TSC1 NM_000368.5 |
c.1972G>A p. Asp658Asn 9:132905606 Exon 15 |
Heterozygous |
35/45.71% |
Autosomal Dominant |
Non-Pathogenic |
|
HBB NM_000518.5 |
c.79G>A p. Glu27Lys 11:5226943 Rs33950507 Exon 1 |
Heterozygous |
17/41.18% |
Autosomal Dominant |
Non-Pathogenic |
|
MYO5A NM_000259.3 |
c.2767A>G p. Ile923Val 15:52372174 Rs757621774 Exon 21 |
heterozygous |
33/45.45% |
Autosomal Recessive |
Non-Pathogenic |
|
CYLD NM_015247.3 |
c.1072T>C p. Leu358= 16:50777875 Rs1270144148 Exon 9 |
Heterozygous |
24/41.67% |
Autosomal Dominant |
Non-Pathogenic |
|
ERCC2 NM_000400.4 |
c.2196_2225del p.Asp732_Leu741del 19:45351687 |
Heterozygous |
21/47.62% |
Autosomal |
Non-Pathogenic |
|
SMARCB1 NM_003073.5 |
C.*594delA 22:23834773 rs35305666 Exon 9 |
Heterozygous |
25/36% |
Autosomal Dominant |
Non- Pathogenic |
Table 2: Whole Exome Sequencing (WES) data of exon-specific mutated genes in DPCs.
Mechanisms of MLH1 in SCs and CRCs as DPCs
MLH1 is one type of mismatch repair gene located on chromosome 3, associated with colorectal cancers (Lynch syndrome) [6]. Defects in MMR proteins give rise to genomic instability, which is characteristic of most cancers. Specifically, 8 – 21% of colorectal cancers are caused by the loss of MLH1 expression [7]. In sporadic CRCs, 10-20% of patients are dMMR, and approximately 95% were due to the promoter methylation of the MLH1 gene. Mutated MLH1 activates PI3/AKT rather than RAS/ MAPK signalling Pathways in CRCs, and it also stimulates other downstream genes such as RAF-MEK-ERK and PI3k/PTEN/AKT signalling Pathways, leading to an increase in the proliferation and progression of Cancerous cells [8].
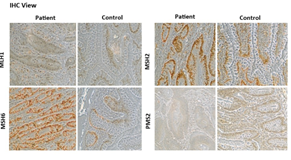
Figure 2: The Immunohistochemistry (IHC) data identified the Major gene (MSI gene) expression between control and dual primary cancer patients.
In the STRING database, we found that MLH1 interacts with other repair genes such as EXO1 and ERCC2. MLH1 heterodimerizes with PMS2 to form MutL-alpha, a component of the postreplicative DNA mismatch repair system (MMR) [9]. DNA repair is initiated by the binding of MutS alpha (MSH2-MSH6) or MutS beta (MSH2-MSH3) to a DNA mismatch, followed by the recruitment of MutL alpha to the heteroduplex [10]. Assembly of the MutL-MutS-heteroduplex ternary complex in the presence of RFC and PCNA is sufficient to activate endonuclease activity of PMS2. It introduces single-strand breaks near the mismatch and thus generates new entry points for the exonuclease EXO1 to degrade the strand [11]. Whereas Exonuclease 1, 5’->3’ doublestranded DNA exonuclease, which may also possess a cryptic 3’>5’ double-stranded DNA exonuclease activity. Functions in DNA mismatch repair (MMR) to excise mismatch-containing DNA tracts directed by strand breaks located either 5’ or 3’ to the mismatch. Also exhibits endonuclease activity against 5’-overhanging flap structures similar to those generated by displacement synthesis when DNA polymerase encounters the 5’-end of a downstream Okazaki fragment [12]. Required for somatic hypermutation (SHM) and class switch recombination (CSR) of immunoglobulin genes [16]. The combined confidence of the functional interaction of MLH1 and EXO1 is 0.999, which denotes a strong correlation between these two genes according to the STRING database.
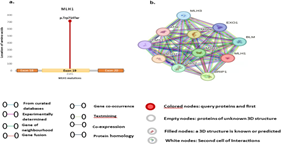
Figure 3: (a). The lollipop data of MLH1, which is a pathogenic gene mutated on Exon 19, and the location of the amino acid p. Trp714Ter. (b). STRING database observed normal MLH1 gene, strongly correlated with other genes like MSH2, PMS2, MSH6, EXO1, and ERCC2.
The role of ERCC2 is a general transcription and DNA repair factor IIH helicase subunit XPD; ATP-dependent 5’-3’ DNA helicase, component of the general transcription and DNA repair factor IIH (TFIIH) core complex, which is involved in general and transcription-coupled nucleotide excision repair (NER) of damaged DNA and, when complexed to CAK, in RNA transcription by RNA polymerase II [13]. In NER, TFIIH acts by opening DNA around the lesion to allow the excision of the damaged oligonucleotide and its replacement by a new DNA fragment [14]. The combined score of EXO1 and ERCC2 is 0.566. Another combined effect of MLH1 and ERCC2 is 0.681, which has a medium correlation between these genes.
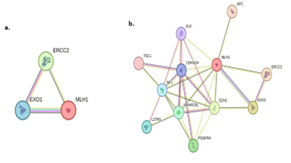
Figure 4: (a). In DPCs, MLH1 mutation has an impact on two other genes, like EXO1 and ERCC2, because they have a strong correlation according to the STRING database. (b). In our study of DPC, we found all these mutated genes; the STRING database represents the role of different exon-specific genes that are strongly correlated with each other.
In this report, we find that when three (MLH1, EXO1, ERCC2) genes are mutated in the cells, it can help to activate the RAS/ MAPK/AKT pathways and stimulate other downstream genes, leading to the proliferation and progression of cancers. In Stomach and Colorectal Cancer, these three mutated genes have a crucial role in the progression of cancer cells and become a malignancy [15].
Chemotherapy for the Treatment of DPCs (SCs and CRCs)
The genome-wide profiling was used to identify common genetic alterations in primary cancers, and the treatment process was also given almost 14 days after surgery for stomach and colorectal carcinoma. The two major drugs, such as Oxaliplatin 180gm and Capecitabine 500gm, are suggested for this patient to recover from the patient’s cancer history and try to inhibit the recurrence rate of cancer progression and proliferation. Oxaliplatin 180 g is a type of cytotoxic Chemotherapeutic drug mainly used in colorectal carcinoma. It is a type of alkylating agent that contains platinum and is used to treat advanced levels of colon cancer [16]. It works by interfering with the DNA replication Process and cross-linking with DNA, stopping the division of Cancer cells and ultimately causing cell death. According to KEGG pathways, oxaliplatin targets genes involved in colon cancer progression, such as those related to the RAS/MAPK pathways, including PDGFR2, ANKRD26, NF1, ALK, and SPTPB. It acts as an inhibitor of the RAS/MAPK pathways, thereby stopping the progression and proliferation of cancer cells.
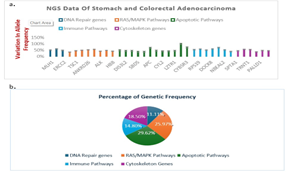
Figure 5: (a). The column Chart depicting the different genes involved in different signalling pathways and percentage of variation in allele frequency (VAF) changes in stomach (SC) and Colorectal Cancers (CRC) as DPCs. (b). Percentage of different signalling pathways specifically mutated genes in Stomach (SCs) and Colorectal cancers (CRCs).
Whereas, Capecitabine 500 mg is a type of antimetabolite chemotherapeutic drug, which is mainly used in stomach carcinoma. when it enters the body, it is converted into fluorouracil, which can directly interfere with the DNA of cancerous cells and inhibit Thymidylate synthase to stop their growth and proliferation [17]. Some clinical observation shows that Capecitabine has immunosuppressive effects, it can induce T-cell apoptosis and produce some anti-inflammatory cytokines, and act as an anticancer drug. Capecitabine increases the stress response in the endoplasmic reticulum and increases the amount of ROS, finally activating mitochondrial-mediated intrinsic apoptotic pathways. However, Capecitabine drugs produce both immunosuppressive and anti-cancer effects.
Discussion
In the present study, common genetic alternation in multiple primary Cancers (SCs & CRC) was observed by genomewide Profiling, providing insight into potential biomarkers. Recent treatments for DPCs remain limited to cisplatin-based chemotherapy, BCG therapy, and surgery, with a median survival of 14-15 months and no recognised second-line therapy [18]. The highest frequency of genetic alternation occurred on chromosomes 3, 16 and 19. Younger patients showed less dependence on genetic factors compared to older patients, suggesting that environmental influences play a role in cancer progression.
Major mutations were detected in DNA repair genes (MLH1, EXO1, ERCC2) and signalling pathways, including RAS/MAPK, apoptotic, cytoskeletal, and immunomodulatory pathways, particularly involving MLH1, EXO1, and ERCC2. MLH1 mutations, frequent in high-grade SCs and CRCs, are closely associated with other repair genes and activate RAS/MAPK signalling, promoting proliferation [19]. Targeted therapies, such as Oxaliplatin and capecitabine, may inhibit the RAS/MAPK Pathways and enhance T-cell-mediated immune responses, respectively.
Epigenetic alterations also suggest novel treatment options. DNA mismatch repair deficiency, especially MLH1 loss, increases EXO1 activity, causing DNA breaks and neoantigen formation. These neoantigens stimulate the dendritic cells and activate tumour-specific T cells, which can recognise and eliminate cancer cells [20]. Although neoantigens have not been reported in DPCs, their discovery could provide new immunostherapeutic targets.
Conclusion
In this report, we highlighted the molecular mechanisms of the major pathogenic gene MLH1 in stomach (SC) and colorectal cancers (CRC). The whole exome sequencing data identified the total number of genes mutated in dual primary cancers. Oxaliplatin and capecitabine are two major drugs that have anticancer properties in these DPCs. The epigenetic alteration of the DNA sequence produces neoantigens, leading to the activation of T cells driven by tumor antigen and killing cancer cells by binding with a specific cell surface receptor. The development of neoantigenspecific drugs will help target treatment options for stomach and colorectal cancers in the future, utilising DPCs.
Acknowledgements
We are grateful to the patient and to his family, who contributed to this study, as well as Dr. C. Mandal, Senior Scientist, who helped to design this study. Additional personnel and funding sources are acknowledged in the supplementary information.
Disclaimer
The author (s) hereby declare that NO generative AI technologies such as Large Language Models (ChatGPT, COPILOT, etc) and text-to-image generators have been used during the writing or editing of this manuscript.
Ethics Approval and Consent to Participate
The Institutional Ethics Committee (IEC) was conducted in Hybrid mode on 14 February 2025 in CNCI.
IEC Ref: CNCI-IEC-JC-2025-14. Finally, the committee approved the ethics. In this ethical approval committee meeting, the consent of the participant member was obtained.
Competing Interests
The authors have declared that no competing interests exist.
List of Authors Contribution
Rabindra Bera: Written and designed the whole manuscript; Sangita Dan: Collected the sample and maintained the entire data of the patient; Soma Sett: Data Curation, conceptualisation; Sparsha Dey: data identification and sample preparation; Palash Dhara: Sample handling and preparation; Krishna Prasad Sahoo: Data Curation, Formal Analysis; Parvez Alam: data interpretation and handling of sample; Jayanta Chakrabarty: Investigation, Maintain a patient’s routine checkup and provide information on treatment options; Subhranshu Mandal: Finalise all the experiments and methods; Sankar Sengupta: Finalise all the experiments and methods; Chandan Mandal: conceived and designed the experiments.
Availability of data and materials
All Project data is available on the Chittaranjan National Cancer Institute (CNCI) website, accessible through the Department of Medical Science’s web browser. https://www.cnci.ac.in
Funding Declaration
Provided by the Chittaranjan National Cancer Institute Core funding agency.
Consent for publication
The authors hereby confirm that all authors have read and approved the final version of the manuscript titled. All authors consent to the publication of this work in the Journal of Scientific Reports, in print and/or electronic form. The authors agree that the work may be edited or formatted to the journal’s requirements without changing the content
References
- Kim HJ, Kim N, Choi YJ, Yoon H, Shin CM, et al. (2016) Clinicopathologic features of gastric cancer with synchronous and metachronous colorectal cancer in Korea: are microsatellite instability and p53 overexpression useful markers for predicting colorectal cancer in gastric cancer patients? Gastric Cancer 19: 798-807.
- Marano L (2023) Dual primary gastric and colorectal cancer: A complex challenge in surgical oncology. World J Gastrointest Oncol, 15: 20492052.
- Li Y, Zhang S, Wang Y, Peng J, Fang F, et al.(2018) MLH1 enhances the sensitivity of human endometrial carcinoma cells to cisplatin by activating the MLH1/c-Abl apoptosis signalling pathway. BMC Cancer 18: 1294.
- Gerlinger L(2020) Immunotherapy Sensitivity of Mismatch RepairDeficient Cancer: Mutation Load Is Not Enough. Cancer Cell 39:16-18.
- Nyqvist J, Kovacs A, Einbeigi Z, Karlsson P, Forssell-Aronsson E, et al.(2021) Genetic alterations associated with multiple primary malignancies. 10: 4465-4477.
- Steinke V, Engel C, Büttne Rr, Schackert H K, Schmiegel W H, et al. (2013) Hereditary Nonpolyopsis Colorectal cancer (HNPCC)/ Lynch Syndrome. 110:32–38.
- Vogt A, Schmid S, Heinimann K, Frick H, Herrmann C,et al. (2017) Multiple primary tumours: challenges and approaches, a review. ESMO Open 2:e000172.
- Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, et al.(2011) Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 305:2304–2310.
- Mahdouani M, Ahmed S B, Hmila F, Rais H, Sghaier R B, et al. (2022) Functional characterisation of MLH1 missense variants unveils mechanisms of pathogenicity and clarifies role in cancer. 17:e0278283.
- Kobayashi H, Ohno S, Sasaki Y, Matsuura M (2013) Hereditary breast and ovarian cancer susceptibility genes (review). Oncol Rep 30:1019– 29.
- Lynch HT, La Chapelle A (2003) Hereditary colorectal cancer. N Engl J Med 348:919–32.
- Zhang C M, Lv J F, Gong L, Yu L Y, Chen X P, et al.(2016) Role of Deficient Mismatch Repair in the Personalized Management of Colorectal Cancer. 13:892.
- Steelman L S, Chappell W H, Abrams S L, Kempf C R, Long J, et al.(2011) Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. 3:192–222.
- Pannafino G, Alani E (2021) Coordinated and Independent Roles for MLH Subunits in DNA Repair. 10:948.
- Hunter N, Borts RH (1997) Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes Dev 11:1573–1582.
- Kunkel TA, Erie D A(2015) Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 49:291–313.
- Tapias A, Auriol J, Forget D, Enzlin J H, Schärer O D, et al. (2004) Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J. Biol. Chem. 279:19074–19083.
- Chen C, Liu H, Li Y,Liu J (2023) Association of ERCC family mutations with prognosis and immune checkpoint inhibitors response in multiple cancers. 13:13925.
- Compe E, Egly JM (2012) TFIIH: When transcription met DNA repair. Nat Rev Mol Cell Biol 13: 343–354.
- Liu J, Hanne J, Britton B M, Bennett J, Kim D,et al.(2016) Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair. Nature 539: 583–587.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.