Switched from Manual to Automated Red Blood Cells Exchange Transfusion: A Retrospective Life Change Improvement from a 12-Year-Old Girl with Sickle Cell Disease in Brazil: A Case Report
by Patricia Gomes Moura*, Fabiana Akil, Leandro Felipe Figueiredo Dalmazzo
Consultant Hematologist, Serum Blood Bank, GSH Group, Brazil
*Corresponding author: Patricia Gomes Moura, Consultant Hematologist, Serum Blood Bank, GSH Group, Brazil
Received Date: 13 August 2025
Accepted Date: 18 August 2025
Published Date: 20 August 2025
Citation: Gomes PM, Akil F, Felipe LFD. (2025). Switched from Manual to Automated Red Blood Cells Exchange Transfusion: A Retrospective Life Change Improvement from a 12-Year-Old Girl with Sickle Cell Disease in Brazil: A Case Report. Ann Case Report. 10: 2380. https://doi.org/10.29011/2574-7754.102380
Abstract
Sickle cell disease (SCD) is a hereditary hemoglobinopathy characterized by recurrent vaso-occlusive crises (VOC), hemolytic anemia, and progressive multi-organ complications. Transfusion therapies, including manual and automated red blood cell exchange (RBCX), remain a standard of care for SCD patients, alongside hydroxyurea (HU).
We describe the clinical course and therapeutic response of a pediatric patient with severe SCD before and after transitioning from manual red blood cell exchange (mRBCX) to automated red blood cell exchange (aRBCX), with a focus on transfusion efficacy, disease burden, and quality of life.
This case highlights the clinical and developmental benefits of aRBCX over mRBCX in a pediatric patient with severe SCD. The findings support the broader use of aRBCX as a disease-modifying approach, particularly in patients with high disease burden and suboptimal response to conventional therapies. Further pediatric-specific studies comparing transfusion modalities are warranted to guide optimized long-term care in SCD.
Keywords: Sickle Cell Disease; Blood Transfusion; Hemoglobinopathy; Automated Red Blood Cells Exchange; Quality of Life.
Introduction
Sickle cell disease (SCD) is an inherited autosomal hematological disorder characterized by a substitution of glutamate by valine in a position 6 of the β-globin gene giving rise to an abnormal hemoglobin or “sickle hemoglobin” (HbS). Under conditions such inflammation, hypoxia or metabolic acidosis, HbS polymerizes, causing red blood cells (RBCs) to take on a characteristic “sickle” shape [1]. SCD includes homozygous hemoglobin S (HbSS), also known as sickle cell anemia. Other isoforms of SCD arise when a different abnormal hemoglobin gene is co-inherited with HbS. These includes HbSC and HbSβ-thalassemia, as well others less common genotype forms such as HbSD [2].
The World Health Organization estimates that the prevalent of SCD worldwide is around 515,000 infants born with the condition each year. Globally, individuals with SCD face premature mortality and debilitating chronic complications that significantly impair their quality of life. Notably, it ranks among the top fifty leading causes of non-communicable deaths worldwide, particularly in the sub-Saharan Africa [1,3]. In Brazil, the Ministry of Health estimates that SCD is present among 60,000 to 100,000 people and 3500 children are born with the condition annually [4].
SCD is associated with life-threatening acute and chronic complications, including chronic hemolytic anemia, acute chest syndrome (ACS), stroke, painful vaso-occlusive crisis (VOC), progressive multiorgan damage, and an increased risk of infection [5,6]. In children, episodes of acute vaso-occlusion crises and stroke are considered the most devastating complications of SCD, leading to frequent crises and hospitalizations. This life-threatening condition results in severe disability, reduced quality of life, increased economic burden, and a high likelihood of increased mortality [6,7].
Among the different therapeutic options to manage the disease, transfusion therapies remain a standard of care for SCD patients, alongside hydroxyurea (HU). These include simple transfusion (ST), manual red blood cell exchange (mRBCX) and automated red blood cell exchange (aRBCX) performed with an apheresis device [8-10].
(Table 1) summarizes the differences between exchanges transfusion modalities.
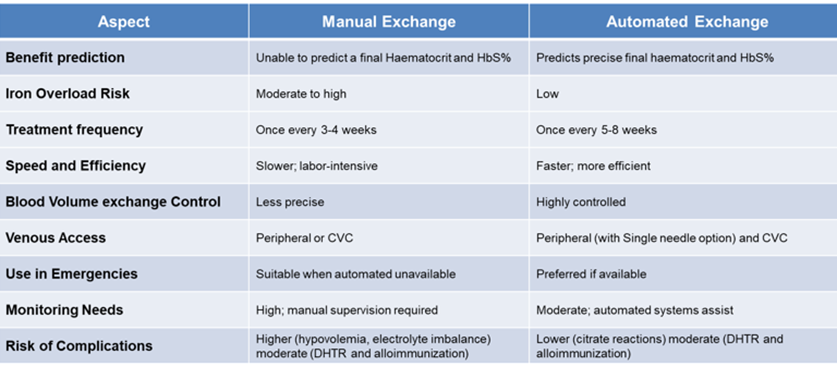
Table 1: Difference between exchange transfusion modalities [8-10, 23, 26-27].
Abbreviations: CVC- central venous catheter; DHTR- Delayed hemolytic transfusion reaction.
The transfusion therapy objectives consist of managing SCD-related complications, by increasing tissue oxygenation and diluting HbS levels below a targeted threshold; improving oxygen-carrying capacity by increasing levels of HbA, decreasing blood viscosity and increasing oxygen saturation by diminishing HbS concentration; and suppressing the production of red blood cells containing HbS by increasing tissue oxygenation [11].
Among the different transfusion techniques, accumulating evidence highlights aRBCX as a faster, more efficient and well-tolerated option, in comparison to ST and mRBCX to removing sickle cells while preserving oxygen-carrying capacity, reducing iron accumulation, and minimizing transfusion-related complications [12-13].
Although the safety and tolerability profile of aRBCX transfusion has become an established and effective therapeutic option for managing SCD [14-16], limited data describing the benefits of automated versus manual RBCX in children, adolescents and young adults have been documented in the literature [14, 17-21].
Here we describe a case of a 12-year-old girl with severe SCD, high episodes of acute VOC and loss of splenic function early in life with an unfavorable clinical outcome besides treatment with manual RBCX transfusion and Hydroxyurea. After been switched to automated RBCX transfusion program, initially aiming at a hematopoietic stem cell transplantation (HSCT), clinical outcomes and quality of life improvements completely changed patient’s life.
Case Presentation
We report on a 12-year-old girl who was diagnosis with homozygous hemoglobin S (HbSS), severe genotype form of SCD. Her retrospective medical files history demonstrate that initial treatment measures were applied to the patient after her birth, such infection prevention with antibiotics and vaccination. During her first two years of life, she had multiple episodes of respiratory infections and severe splenic sequestration crises managed with broad-spectrum antibiotics and intravenous fluids. One of these events, her hemoglobin presentation was 4.8 g/dL, and she was given a simple transfusion. Due to recurrent splenic sequestration crises, she underwent to a splenectomy at the age of 2.5 years.
The patient neurologic function for stroke risk assessment was periodically monitored by conventional transcranial doppler (TCD) and at age of 5-years old the patient had an abnormal ultrasound result showing a Bifurcation of the Internal Carotid Artery (BIFD) 203 and Right Middle Cerebral Artery (MCA) 188. The patient’s severe form of SCD, with constant respiratory and acute neurological complications, indicated immediate inclusion in the chronic manual RBCX transfusion program and daily intake of Hydroxyurea with an initial dose of 12 mg/kg of body weight. One year later, her ultrasound result (TCD) showed a Low conditional result with a cerebral blood flow velocity of 179 cm/s. During this period of treatment, clinical improvement was minimal, and Hydroxyurea dosage increased to 20 mg/kg of body weight. From 2020 up to May-2023 her historic medical files showed that the patient had a high number of emergency visits and hospitalizations due to VOCs, totaling 24 episodes during this time. A notable hospitalization was due to a transient ischemic attack (TIA), underscoring the severity of the disease course despite optimized Hydroxyurea and manual RBCX management.
Data from manual transfusion therapy monitoring showed limited efficacy in reducing hemoglobin S (HbS%) (Figure 1A) to the target threshold recommended (˂30%), based on ASFA, ASH and the Brazilian recommendation Guidelines [22-24]. During this period, she had a history of allergic reactions, requiring pre-medication prior to transfusions. Iron chelation therapy has been part of her treatment regimen, considering her increased levels of ferritin (Figure 1B). Given the suboptimal response to the manual transfusion therapy, high disease burden and neurological risk, the patient was recommended to be enrolled in the hematopoietic stem cell transplantation (HSCT).
In May 2023, the patient was recommended by her previous medical center to our Hemotherapy service at Serum Blood Bank – GSH Group to receive adequate blood transfusion support to undergo HSCT. It has been well described in the literature that reducing the HbS% to less than 30% is recommended before the start of HSCT, therefore the pre-HSCT blood transfusion can be beneficial by modulating the marrow environment to improve engraftment [25]. It is important to note that Serum Blood Bank – GSH Group services prioritize donor selection using extended red cell antigen profile phenotyping to ensure compatibility with matched donors for all patients under transfusion program. All RBCs prior to transfusion are leukocytes depleted, phenotypically matched for D, C/c, E/e, K/k, antigens and antigen-negative for alloantibodies. It is critical to obtain a patient’s antibody history from other services that provided prior transfusions, however, since the patient was switched to our service, no alloimmunization profile has been identified (data not shown).
From June 2023 till date, the patient underwent a chronic transfusion regimen with automated red blood cell exchange performed using the Spectra Optia TM Apheresis Systems version 12.0 software (Terumo Blood and Cell Technologies, Lakewood, CO). A retrospective comparative analysis between manual and automated exchanges procedures in pediatric patients demonstrated the ability of aRBCX to rapidly remove RBCs containing HbS and replace with normal HbA RBCs from health compatible donors, achieve HbS% and Hematocrit (Hct%) targets and manage blood viscosity and iron overload [14, 17-19]. Consistent with this, the patient aRBCX data demonstrated that the number of pRBC units’ utilization per procedure able to achieve the target HbS% ˂ 30% was 3 ± 0.5 (mean ± SD), the post-procedure final FCR% (fraction of remaining cells) was 49 ± 6.1 (mean ± SD) and pre- and post-procedure hematocrit (%) were 24.4 ± 1.5 and 30 ± 1.3 (mean ± SD), respectively. Patient venous access for aRBCX procedures were performed via peripheral catheter using a single needle (AV fistula 16g) that can alternate blood withdrawal and return [26]. Although the procedure time can be slightly longer when compared to the usual double needle, the average time for the patient procedures was 49 ± 9.8 (min; mean ± SD). All the details of the aRBCX procedures are summarized in (Table 2)
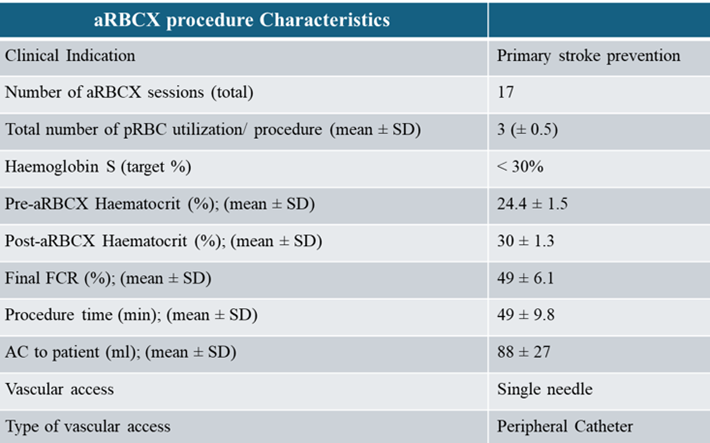
Table 2: Details of the automated Red Blood Cell Exchange Procedures.
The patient experienced retrospectively from 2020 up to date, 46 exchange transfusion procedures (29 manually and 17 automated performed). The mean post-exchange HbS% levels were significantly lower in aRBCX (23.6% vs 42.6%, p ˂ 0.0001) versus manual exchange. In the aRBCX, the HbS% was reduced to ˂ 30% and maintained with a mean pre-exchange HbS of 48.8% between procedures (Figure 1A).
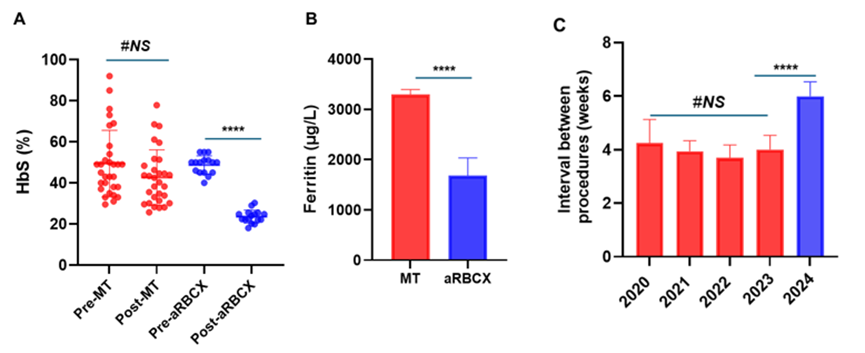
Figure 1: Comparison of clinical and laboratory outcomes between manual (mRBCX) and automated red blood cell exchange (aRBCX) in a pediatric sickle cell disease patient. (A) Hemoglobin S (HbS%) levels, measured by High Performance Liquid Chromatography (HPLC), pre- and post-exchange transfusion aRBCX significantly reduced post-exchange HbS% compared to mRBCX (**** One-way ANOVA with Tukey’s multiple comparison test; p < 0.0001); (B) Serum ferritin levels during the mRBCX period (red) versus aRBCX period (blue), indicating improved iron overload control after switching to automated exchange (**** Unpaired two tailed t-test p < 0.001); (C) Interval between transfusion procedures in weeks. aRBCX allowed significantly extended time between procedures compared to mRBCX (**** One-way ANOVA with Tukey’s multiple comparison test; p < 0.0001). Data are represented as mean ± SD or individual values where indicated. Statistical significance was determined using One-way ANOVA with Tukey’s multiple comparison test and unpaired two tailed t-tests. Red bars/dots represent manual exchange; blue bars/dots represent automated exchange.
The limitations of manual transfusion exchange as the inability to predict final hematocrit and exchange the adequate blood volume to diminish HbS to ˂ 30%, was translated into the worse clinical patient outcome period. Since the patient underwent aRBCX, she has shown excellent clinical progress, with significant physical development, gaining 15 kg in weight and growing 17 cm in height, with a Body Mass Index (BMI) compatible to children at same age and sex as determined by the growth chart of World Health Organization (WHO) (Figure 2). The serum ferritin levels decreased from 3297 µg/L during the manual exchange to 1680 µg/L in the aRBCX period (Figure 1B), indicating improved iron overload management. Important to note that the interval between procedures increased from 4 ± 0.8 to 6 ± 0.9 weeks (mean ± SE) (Figure 1 C).
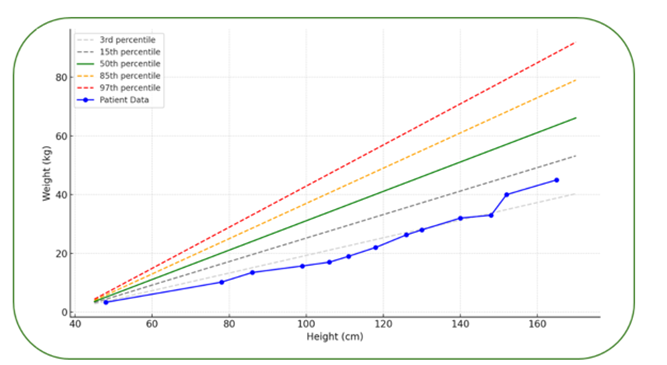
Figure 2: Weight-for-Height Percentile Chart with WHO reference percentiles fitted with the patient’s growth trajectory from birth to age 12, represented by the blue curve.
The patient clinical outcomes were evaluated through the analysis of hospitalization associated with the period of VOC when the patient underwent manual exchange versus aRBCX over the period from 2020 up to date. During manual exchange transfusion timeframe, the patient experienced a significantly higher burden of clinical complications related to VOC. Patient underwent 16 visits to Accident & Emergency (A&E), 8 hospital admissions, and a total of 29 days spent in hospital due to VOC-related complications. During the aRBCX period, the patient had no recorded A&E visits, no hospitalizations and no inpatient days attributable to VOC as shown in (Table 3).
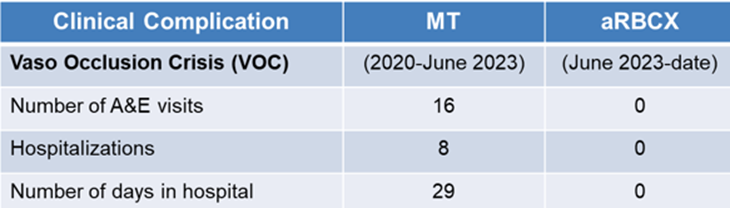
Table 3: Patient clinical complications records during the exchange transfusion treatment.
Abbreviations: MT-manual transfusion; aRBCX-automated red blood cells exchange.
Overall, the patient has responded well to automated transfusion therapy, showing positive clinical and developmental outcomes. Due to her clinical improvement, she has not compliance with Hydroxyurea and the iron chelation agent for the last 8 months. The family is not considering HSCT possibilities anymore. A cartoon representative summarizes the timeline of the patient undergoing treatment (Figure 3).
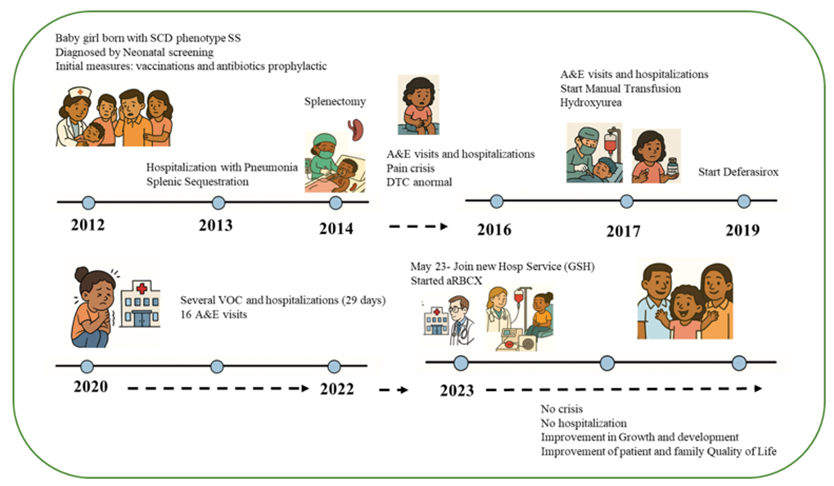
Figure 3: Representative case description cartoon: Girl’s Journey with Sickle Cell Disease: “Growing Stronger”.
Discussion
Sickle Cell Disorder (SCD) is a complex inherited hematological blood disorder affecting millions globally, including a significant population in Brazil [4] SCD leads to various acute and chronic complications, including acute chest syndrome (ACS), stroke, painful vaso-occlusive crisis (VOC), progressive multiorgan damage, and an increased risk of infection. This common, life-threatening condition results in severe disability, reduced quality of life, increased economic burden, and a high likelihood of increased mortality [5-7].
This case illustrates the clinical impact of transitioning from manual red blood cell exchange (mRBCX) to automated red blood cell exchange (aRBCX) in a child with severe sickle cell disease (SCD). Despite early diagnosis and aggressive disease-modifying interventions, including Hydroxyurea and chronic mRBCX, the patient continued to experience frequent vaso-occlusive crises (VOCs) and risk of neurologic complications.
A critical limitation in her prior management was the consistent failure of mRBCX to reduce hemoglobin S (HbS%) to below the recommended therapeutic threshold of 30%, as recommended by ASFA, ASH and the Brazilian guidelines [22-24]. This suboptimal control of HbS% likely contributed to the ongoing disease burden resulting in frequent emergency visits, hospitalizations, and the transient ischemic attack (TIA) experienced despite regular transfusion support. The unpredictable efficacy of mRBCX, due in part to its manual nature, limited exchange volume, high risk of complications such as electrolyte imbalance, hypovolemia, hemodynamic instability and inability to precisely control post-procedure HbS% and hematocrit ratio resulted in an overall insufficient therapeutic response [12, 14, 18, 19, 27-29].
Several studies have demonstrated the benefits of aRBCX in SCD pediatric patients, including prevention of primary stroke, recurrent stroke, reduced hospitalization for vaso-occlusive crisis, and acute chest syndrome [17-25]. Consistent with this, the initiation of aRBCX in the patient led to a rapid and sustained reduction of HbS% to <30%, with significantly improved control between procedures (mean pre-exchange HbS% of 48.8% and post-exchange HbS% of 23.6%). This effective suppression of sickle hemoglobin was accompanied by a complete resolution of VOC episodes, elimination of emergency and inpatient admissions and improved interval between procedures. A previous study has demonstrated the ability of aRBCX to lower ferritin levels [19] under chronic transfusion treatment; this aligns with the present case report that shows a notable reduction in iron overload, as evidenced by decreasing ferritin levels.
The precision and efficiency of aRBCX enabled by automated control of exchange volumes, hematocrit, and HbS targets make it a clearly superior modality in achieving disease control in high-risk pediatric SCD patients. Importantly, clinical stability led the family’s patient to defer consideration of hematopoietic stem cell transplantation (HSCT), further highlighting the quality-of-life benefits of well-managed transfusion therapy.
It is important to highlight that the concomitant use of Hydroxyurea (HU) and chronic blood transfusion therapy in patients with SCD remains a topic of constant debate, with conflicting evidence regarding their combined efficacy. Hydroxyurea has demonstrated significant benefits in reducing the frequency of vaso-occlusive crises, acute chest syndrome, and transfusion needs by increasing fetal hemoglobin (HbF) levels [30, 31]. However, when used alongside chronic transfusion red blood cell exchange, the additive value becomes unclear. Previous reports suggest that combining HU with transfusions may offer synergistic effects by reducing overall HbS production and inflammation, while others report minimal additional benefits and highlight concerns regarding increased treatment burden, adherence challenges, and unclear impact on transfusion volume or HbS suppression [32, 33].
In this case, despite dose escalation of HU, the patient continued to experience high disease burden and suboptimal control of HbS% while on manual RBCX, suggesting limited benefit of HU under inadequate transfusion management. After switch transitioning to automated RBC exchange, which successfully suppressed HbS levels and resolved clinical complications, the patient discontinued HU without recurrence of VOCs or other complications, further underscoring the need for individualized treatment decisions and a better understanding of when dual therapy is warranted.
The patient's recent discontinuation of Hydroxyurea and iron chelation therapy without medical consent raises important concerns about long-term disease management and the importance of sustained multidisciplinary care and adherence.
It is important to emphasize the great need to report the clinical outcomes of aRBCX programmes in the pediatric settings, given the limited data availability of this exchange modality nationwide. By sharing these findings, we can demonstrate the importance of this treatment option in diverse healthcare systems not only in Brazil but also in other countries.
Conclusion
In summary, this case strongly reinforces the limitations of manual exchange transfusion in achieving adequate HbS suppression and the superior efficacy of automated exchange in both biochemical and clinical outcomes. For pediatric patients with severe SCD and high disease burden, transitioning to aRBCX should be considered early to optimize disease control, reduce complications, and improve long-term prognosis.
Most notably, the switch to aRBCX led to a dramatic improvement in the patient’s and family quality of life, marked by complete resolution of hospitalizations, elimination of painful crises, uninterrupted school attendance, and restored participation in daily childhood activities, transforming both her clinical trajectory and social well-being.
Informed consent: Written informed consent was obtained from the patient’s parents.
Acknowledgements: The authors thank Terumo BCT for their support in the training and implementation of the aRBCX at GSH, as well Dr Leonardo Moraes from Terumo BCT for the research support to this case-study.
Author contributions: Dr Patricia Gomes Moura, Dr Fabiana Akil and Dr Leandro Felipe Figueiredo Dalmazzo contributed to the data collection, draft and final version of this report.
Conflict of interest: The authors declare no conflicts of interest.
References
- Piel FB, Steinberg MH, Rees DC. (2017) Sickle Cell Disease. N Engl J Med. 376: 1561-1573.
- Ware RE, De Montalembert M, Tshilolo L, Abboud MR. (2017) Sickle Cell Disease. Lancet. 390: 311-323.
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, et al. (2013) Global Epidemiology of Sickle Haemoglobin in Neonates: A Contemporary Geostatistical Model-Based Map and Population Estimates. Lancet. 381: 142-151.
- Telles L, Melo PHM, Dornelas LB, Lech GE, Sampaio NZ, et al. (2025) Epidemiological Profile Trends and Cost of Pediatric Sickle Cell Disease in Brazil From 2008 to 2022. J Pediatr (Rio J). 101: 110-116.
- Rees DC, Williams TN, Gladwin MT. (2010) Sickle-Cell Disease. Lancet. 376: 2018-2031.
- Elendu C, Amaechi DC, Alakwe-Ojimba CE, Elendu TC, Elendu RC, et al. (2023) Understanding Sickle Cell Disease: Causes, Symptoms, and Treatment Options. Medicine (Baltimore). 102: e35237.
- Lobo C, Moura P, Fidlarczyk D, Duran J, Barbosa R, et al. (2022) Cost Analysis of Acute Care Resource Utilization Among Individuals With Sickle Cell Disease in a Middle-Income Country. BMC Health Serv Res. 22: 42.
- Brandow AM, Liem RI. (2022) Advances in the Diagnosis and Treatment of Sickle Cell Disease. J Hematol Oncol. 15: 20.
- Sharma D, Ogbenna AA, Kassim A, Andrews J. (2020) Transfusion Support in Patients With Sickle Cell Disease. Semin Hematol. 57: 39-50.
- Abboud MR. (2020) Standard Management of Sickle Cell Disease Complications. Hematol Oncol Stem Cell Ther. 13: 85-90.
- Davis BA, Allard S, Qureshi A, Porter JB, Pancham S, et al. (2017) Guidelines on Red Cell Transfusion in Sickle Cell Disease Part II: Indications for Transfusion. Br J Haematol. 176: 192-209.
- Al Mozain N, Elobied Y, Al-Omran A, Aljaloud A, Omair AB, et al. (2023) Comparative Study Between Chronic Automated Red Blood Cell Exchange and Manual Exchange Transfusion in Patients With Sickle Cell Disease: A Single Center Experience From Saudi Arabia. Asian J Transfus Sci. 17: 91-96.
- Kelly S. (2023) Logistics, Risks, and Benefits of Automated Red Blood Cell Exchange for Patients With Sickle Cell Disease. Hematology Am Soc Hematol Educ Program. 2023: 646-652.
- Duclos C, Merlin E, Paillard C, Thuret I, Demeocq F, et al. (2013) Long-Term Red Blood Cell Exchange in Children With Sickle Cell Disease: Manual or Automatic? Transfus Apher Sci. 48: 219-222.
- Quirolo K, Bertolone S, Hassell K, Howard T, King KE, et al. (2015) The Evaluation of a New Apheresis Device for Automated Red Blood Cell Exchange Procedures in Patients With Sickle Cell Disease. Transfusion. 55: 775-781.
- Tsitsikas DA, Sirigireddy B, Nzouakou R, Calvey A, Quinn J, et al. (2016) Safety, Tolerability, and Outcomes of Regular Automated Red Cell Exchange Transfusion in the Management of Sickle Cell Disease. J Clin Apher. 31: 545-550.
- Koehl B, Sommet J, Holvoet L, Abdoul H, Boizeau P, et al. (2016) Comparison of Automated Erythrocytapheresis Versus Manual Exchange Transfusion to Treat Cerebral Macrovasculopathy in Sickle Cell Anemia. Transfusion. 56: 1121-1128.
- Escobar C, Moniz M, Nunes P, Abadesso C, Ferreira T, et al. (2017) Partial Red Blood Cell Exchange in Children and Young Patients With Sickle Cell Disease: Manual Versus Automated Procedure. Acta Med Port. 30: 727-733.
- Dedeken L, Le PQ, Rozen L, El Kenz H, Huybrechts S, et al. (2018) Automated RBC Exchange Compared to Manual Exchange Transfusion for Children With Sickle Cell Disease is Cost-Effective and Reduces Iron Overload. Transfusion. 58: 1356-1362.
- Elenga N, Vantilcke V, Martin E, Cuadro E, Selles P, et al. (2022) Red Blood Cell Exchange in Children With Sickle Cell Disease. Int J Hematol. 115: 107-113.
- Van Hattem J, Maes P, Esterhuizen TM, Devos A, Ruppert M, et al. (2023) Erythrocytapheresis in Children and Young Adults With Hemoglobinopathies and Iron Overload in Need of Iron Chelation Therapy. J Clin Med. 12: 6287.
- Connelly-Smith L, Alquist CR, Aqui NA, Hofmann JC, Klingel R, et al. (2023) Guidelines on the Use of Therapeutic Apheresis in Clinical Practice - Evidence-Based Approach From the Writing Committee of the American Society for Apheresis: The Ninth Special Issue. J Clin Apher. 38: 77-278.
- Chou ST, Alsawas M, Fasano RM, Field JJ, Hendrickson JE, et al. (2020) American Society of Hematology 2020 Guidelines for Sickle Cell Disease: Transfusion Support. Blood Adv. 4: 327-355.
- Loggetto SR, Veríssimo MPA, Darrigo-Junior LG, Simões RDS, Bernardo WM, et al. (2022) Guidelines on Sickle Cell Disease: Primary Stroke Prevention in Children and Adolescents. Hematol Transfus Cell Ther. 44: 85-94.
- Zheng Y, Chou ST. (2021) Transfusion and Cellular Therapy in Pediatric Sickle Cell Disease. Clin Lab Med. 41: 101-119.
- Kearney L, Bosnick R, Phillips H, Ghio A, Cullen D, et al. (2024) Optimization of Single-Needle Red Cell Exchange in Patients With Sickle Cell Disease. J Clin Apher. 39: e22118.
- Bendiola A. (2022) Improving Outcomes for Children and Young People With Sickle Cell Disease. Nursing Times. 118: 12.
- Kuo KH, Ward R, Kaya B, Howard J, Telfer P. (2015) A Comparison of Chronic Manual and Automated Red Blood Cell Exchange Transfusion in Sickle Cell Disease Patients. Br J Haematol. 170: 425-428.
- Wallace LR, Thibodeaux SR. (2022) Transfusion Support for Patients With Sickle Cell Disease. Transfus Apher Sci. 61: 103556.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, et al. (2014) Management of Sickle Cell Disease: Summary of the 2014 Evidence-Based Report by Expert Panel Members. JAMA. 312: 1033-1048.
- Kanter J, Kruse-Jarres R. (2013) Management of Sickle Cell Disease From Childhood Through Adulthood. Blood Rev. 27: 279-287.
- Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, et al. (2016) Hydroxycarbamide Versus Chronic Transfusion for Maintenance of Transcranial Doppler Flow Velocities in Children With Sickle Cell Anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): A Multicentre, Open-Label, Phase 3, Non-Inferiority Trial. Lancet. 387: 661-670.
- Nickel RS, Margulies S, Panchapakesan K, Chorvinsky E, Nino G, et al. (2024) Adding Hydroxyurea to Chronic Transfusion Therapy for Sickle Cell Anemia Reduces Transfusion Burden. Transfusion. 65: 38-49.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.