Pseudohypoaldosteronism Syndrome, Respiratory Manifestations; Functional-Morphological and Genetic Aspects of the Mucociliary System
by V.I. Kobylyansky*
Federal State Budgetary Institution Research Institute Of Pulmonology Of The Federal Medical And Biological Agency of Russia, Moscow, Russia
*Corresponding Author: Kobilyansky Vyacheslav Ivanovich, The Educational Center of the Research Institute of Pulmonology of the Federal Medical and Biological Agency of Russia, Moscow, Russia
Received Date: 14 June 2025
Accepted Date: 20 June 2025
Published Date: 23 June 2025
Citation: Kobylyansky VI (2025) Pseudohypoaldosteronism Syndrome, Respiratory Manifestations; Functional-Morphological and Genetic Aspects of the Mucociliary System. J Surg 10: 11355 https://doi.org/10.29011/2575-9760.011355
Abstract
The physical properties of the mucous coating of the respiratory tract epithelium, which is an important link in the Mucociliary System (MCS), the function of which is mucociliary clearance - the leading protective mechanism of the lungs, are regulated to a large extent by ion transport in the epithelium. The dominant characteristic of the latter is the activity of sodium absorption in Epithelial Sodium Channels (ENC), the disturbances of which play a significant pathogenetic role in various lung diseases. And if the state of the MCS has been studied to a large extent and reflected in such a pathology as cystic fibrosis, in which ENC disorders play a cardinal role in the pathogenesis of its pulmonary manifestations, then this aspect in Pseudohypoaldosteronism (PHA), which is also determined by ENC and MCS disorders, but of a different nature, has been studied and reflected in the literature far from sufficiently. Information about PGA in domestic medicine is extremely limited, and pulmonological aspects and data on the state of the MCS in PGA are present only in a small number of works by foreign authors and are absent in domestic medicine.
The aim: of the work is to expand the scientific and practical content on the etiopathogenetic and therapeutic aspects of PGA from the perspective of respiratory manifestations and the mucociliary system.
Research Methodology: An analysis of 31 full-text literary sources was carried out, selected based on the results of a search in biomedical scientific information databases, including Index Medicus, PubMed, Embase, Cohrane.
It was established that the state of the ENC in PHA plays a fundamental role in the balanced work of the main links of the MCS. It was determined that in patients with primary systemic PHA, mutations in the genes of the ENC subunits cause loss of their function and absence of sodium absorption from the airway surfaces, causing a significant increase in the volume of fluid on the airway surface and respiratory symptoms. Data on the state of the MCS function are contradictory, which requires clarification.
Conclusion: Sodium transport by the ENC of the respiratory tract plays a significant role in the regulation of their mucous coating. In primary systemic PHA, it is impaired, which contributes to a decrease in fluid absorption and an increase in the volume of the mucous coating, which can manifest itself clinically. Data on the effect of this on the MCS function are contradictory, which requires adequate clarification by its direct assessment in vivo using a standardized method and taking into account the influence of various factors.
Keywords: Blockade; Epithelial Sodium Channels; Hypersecretion; Mucociliary System; Pseudohypoaldosteronism
Introduction
The earliest and most significant protective mechanism of the respiratory system is the function of the Mucociliary System (MCS), Mucociliary Clearance (MCC), the disorders of which are involved in the pathogenesis of various pathologies. This function largely depends on the physical properties, including the volume, composition and rheology, of an important link in the MCS - the mucous coating of the Respiratory Tract (RT) epithelium, interacting with its motor link - the ciliary apparatus. The physical properties of the mucous coating are regulated to a large extent by ion transport in the epithelium, the dominant characteristic of which is the activity of sodium absorption in the Epithelial Sodium Channels (ESC). Various disorders of sodium transport have been described, indicating its role in the function of the MCS, in particular in cystic fibrosis, in which ENC is activated and excess sodium absorption occurs, leading to dehydration of the mucous coating and disruption of the MCC [1]. There is also a condition when the ENC is blocked and sodium absorption slows down or is absent, contributing to fluid retention and an increase in the volume of DP mucus, which can manifest itself in respiratory symptoms, as is the case with the so-called Pseudohypoaldosteronism (PHA), which is hereditary [2]. This disease is characterized by a violation of sodium transport in epithelial cells and a loss of salt and is caused by a decrease in the sensitivity of the effector organs to the action of aldosterone, the synthesis of which is not impaired. Undoubtedly, the expansion and deepening of our understanding of the mechanisms of the MCS, their breakdowns increases the possibilities for improving the early diagnosis and treatment of respiratory pathology. And if the state of the MCS has been studied to a large extent in CF, then in PHA there are isolated foreign works and there is practically no coverage in domestic medicine concerning the pulmonary aspects of this genetically determined pathology, and especially from the position of the MCS, which was the purpose of this work. Material and methodsA selection and subsequent analysis of publications posted on the PubMed, Embase, Cochrane, Index Medicus platforms was carried out using key words and phrases, including pseudohypoaldosteronism, epithelial sodium channels, blockade, hypersecretion, mucociliary system function. Of the relatively small number of works that are available in the literature, 31 full-text literature sources were included in the analysis with narrowing at each stage, and then the selected data were structured and used to prepare a systematic review (Figure 1).
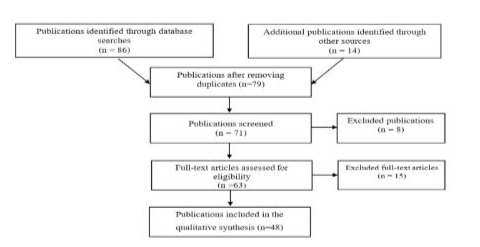
Figure 1: Scheme of publication selection.
Brief introduction to PGA, clinical symptoms, pathogenetic mechanisms
PGA is a genetically determined syndrome of a heterogeneous group of diseases, which is characterized by impaired sodium transport in epithelial cells and salt loss and is caused by a decrease in the sensitivity of effector organs to the action of aldosterone, the synthesis of which is not impaired. PGA syndrome was first described in the middle of the last century in children in the first months of life [3]. This is a group of rare disorders, including mainly diseases of genetically determined genesis, characterized by resistance to the action of aldosterone. In this case, hyponatremia, hyperkalemia and increased plasma renin activity against the background of increased aldosterone concentrations occur. There are two main types of PGA, which have significant differences in etiology and manifestations. PGA type 2 is inherited in an autosomal dominant manner and is known as familial hyperkalemic hypertension or Gordon syndrome. It is characterized by abnormalities in the body's regulation of sodium and potassium levels and is caused by mutations in the serine-threonine kinase genes WNK4, WNK1, KLHL3, and CUL3, which are involved in the regulation of sodium transport in the kidneys, in the distal convoluted tubule, and the control of sodium and chloride levels in the body. Mutations in these genes cause excessive reabsorption of sodium and chloride, which ultimately leads to hyperkalemia. There is an increase in blood volume regardless of normal or low aldosterone levels due to increased activity of sodium transporters in the kidneys. Clinically, hyperkalemia develops first, with hypertension appearing later in life. Nonspecific symptoms such as nausea, vomiting, severe fatigue, muscle weakness, and hypercalciuria may also be observed. It is assumed that epileptic manifestations may occur due to potassium surges leading to abnormal activity of CNS neurons. Given the lack of relevance of this type of PGA to the problem under consideration, we will not dwell on it in detail. PGA Type 1 (PGA1) is characterized by the body's inability to adequately respond to aldosterone, a hormone that is critical for regulating electrolyte levels and is the result of mutations in the Epithelial Sodium Channel (ESC) in any of the three subunits (α, β, γ) of the amyloid-sensitive ENC. This condition often manifests itself as dehydration, since the kidneys have difficulty retaining enough sodium, which leads to symptoms such as increased thirst and dry mouth. PGA1, by disrupting the electrolyte balance, leads to low sodium and high potassium in the blood. PGA1 is a heterogeneous disease, since it is caused by mutations in different genes. On the one hand, these are mutations in the NR3C2 gene (encodes the mineralocorticoid receptor), which cause the synthesis of a non-functional receptor that cannot bind aldosterone (or function correctly), affecting the distal cells of the nephron and playing an important role in the regulation of sodium and potassium homeostasis. On the other hand, PGA1, inherited in an autosomal recessive manner, is caused by mutations in both alleles of the SCNN1A, SCNN1B or SCNN1G genes, encoding various subunits of the ENC, which is responsible for sodium reabsorption and potassium secretion. In turn, PGA1 can be divided into primary and secondary or acquired. In the latter case, signs of hypoaldosteronism usually arise as a result of organic or functional obstructive and/or infectious uropathy, side effects of drugs, including cyclosporines, beta-blockers, potassium-sparing diuretics, etc., and tubulointerstitial diseases, which also do not fall within the scope of the problem under consideration. As for primary PGA1, two clinically different forms are distinguished - systemic and renal [4].
The systemic form is inherited in an autosomal recessive manner and is caused by the loss of salt as a result of mutations in the genes that control ENC in the tissues of a number of organs, including the lungs, kidneys, colon, salivary glands, and sweat ducts. Under the influence of these mutations, ENC are blocked and there is no sodium absorption, which contributes to water retention on the surface of the respiratory tract [5]. The renal form is inherited in an autosomal dominant manner and is caused by mutations in the gene of mineralocorticoid receptors of the renal tubules. It is characterized by loss of salt in the kidneys. Both forms of PGA1 can be present in the first week of a child's life, accompanied by dehydration, natremia, hyperkalemia. We are more interested in systemic PGA1, otherwise Autosomal Recessive PGA1 (ARPGA1), given its possible manifestations in the bronchopulmonary system, requiring correction, the possibilities of which are in short supply and require appropriate developments.It has been observed that impaired ENC activity causes a significant decrease in fluid transport in the RT in neonates at birth, its excess in the lungs and the inability to clear it, which can lead to death in the neonatal period. It has been established that this phenotype of the disease usually involves missense mutations that cause impaired ENC function and can be eliminated by its expression [6,7]. An increase in the volume of fluid on the RT surface is, to a certain extent, the opposite of the pathophysiological mechanisms that manifest themselves through ENC activation and, conversely, increased absorption, which occur in CF [8]. Children with the systemic form of ARPGA1 often suffer from recurrent lower RT diseases already during the first years of life [9]. Genetic defects in the ENC alpha subunit are associated with pronounced pulmonary symptoms [10]. They have identified structural changes or stop mutations in the genes for the ENC subunits on both alleles, which are believed to impair its function [11]. It is assumed that patients with this form of PGAS, but without identified mutations, could have intronic or promoter mutations or mutations affecting other proteins responsible for the assembly and function of ENC. Clinical pulmonary manifestations vary significantly - from severe, occurring more often before the age of 2, to asymptomatic, which are more common in adults with the systemic form of the disease [12-14].
More frequent and severe symptoms in ARPGA1 than in adults, manifested by cough, shortness of breath, wheezing (which can be mistaken for bronchial asthma), often the development of an infectious process, are noted in children of early years of life. This may be associated with a small RD diameter, since it predisposes to greater RD obstruction due to an increased volume of periciliary fluid and significant dilution of surfactant, which ensures stabilization of the RD lumen [15,16]. In one of the studies, all patients had pulmonary symptoms of varying severity. The results of a bacterial study to some extent resembled those in cystic fibrosis, but the development of chronic lung disease and progressive decline in lung function were not observed. Genetic deficiencies of the alpha subunit of ENC are associated with severe pulmonary symptoms, which, however, clearly differ from the symptoms in cystic fibrosis [10]. There are studies in which patients with PHAS, along with increased sweating and an increase in the concentration of salt in saliva, noted an increase in the frequency of RD infection [17,18]. However, a comprehensive study did not reveal chronic RD infection and / or the presence of bronchiectasis, which would give reason to believe about a violation of the MCC. On the contrary, there are data indicating that there may be an increase in the MCC velocity in ARPGA1 [19]. This may indicate a compensatory increase in the MCC function, allowing the removal of excess volume of DP secretion. This assumption is consistent with one of the hypotheses that the upper viscous layer of the mucous coating (gel) is transported due to the movement of the lower liquid layer (sol), provided by the work of the ciliary apparatus. This refutes another hypothesis that the gel is moved due to its pushing by the claws of the cilia, since in this case the MCC velocity in PGAS (contrary to the data obtained) would decrease due to the lack of contact of the cilia with the gel. However, accelerated clearance may also be the result of an improvement in the impaired rheological properties of the gel under the influence of an increase in the sol volume, and then the above-mentioned confirmation of the hypothesis, like the hypothesis itself, loses its meaning [20]. Perhaps, as a result of this, the disease often proceeds asymptomatically. Therefore, the opinion that an increase in the periciliary layer slows down the transport of sputum is to some extent controversial. This is also indicated by the data of the meta-analysis. In fact, the question of the criticality of the periciliary, liquid layer, sol, for the function of the MCS is more complex and depends on a number of factors, including the transmembrane transport of liquid, regulated in many ways by protein structures called aquaporins, and on the state of the large gene apparatus that supervises all the structures of the MCS, especially those related to its main links, etc [21].
Some critical aspects of the concepts of ARPGA from the position of the CMC
At the same time, it should be taken into account that an increase in the amount of bronchial contents and an increase in the thickness of the periciliary layer of the mucous coating or shortening of cilia, which contributes to a change in the interaction of the leading links of the CMC, contribute to a decrease in the efficiency of the leading link of the CMC, the ciliary apparatus and, accordingly, a decrease in the effectiveness of the CMC [22]. At the same time, the loss of length of cilia, contributing to the disruption of the CMC, can be associated with changes in specialized intracellular intraflagellar transport responsible for the formation and maintenance of the structure and function of cilia [22,23]. This is clearly indicated by the results of a number of studies. In particular, it has been clearly demonstrated in smokers that a morphologically confirmed shortening of the cilia of the AP by 15% leads to a decrease in the CMC, which plays an important role in the pathogenesis of lung diseases caused by smoking [24]. And this is not surprising, since the length of the cilia is comparable to the thickness of the periciliary layer and, thus, the claws optimally contact the gel layer in order to effectively push it. Naturally, shortening of the cilia and/or thickening of the periciliary layer or significant liquefaction of the gel will reduce this interaction even with a normal ciliary structure and activity of the cilia. In this case, the tips of the cilia will not reach the gel layer and push it towards the nasopharynx or, reaching it, will not receive sufficient contact and friction with the upper layer of mucus, and the layer itself will be too heterogeneous for its movement, i.e. the work of the cilia will be idle and ineffective in transporting the gel, contributing to its decrease, and, consequently, the MCC. Therefore, a significant increase in the average mucociliary transport velocity, assessed in one of the studies in only 3 patients, compared with healthy subjects, against the background of a state of hypersecretion, which, as a rule, contributes to the violation of the MCC and is compensated as a result by cough clearance, most likely does not reflect the real state of the MCC function, not fitting into the existing ideas about it [19]. The analysis showed that, obviously, there could be factors contributing to a more proximal deposition of the inhalant relative to healthy individuals, overestimating the MCC indicators. These include the influence of bronchial obstruction caused by both hyperreactivity, which is indicated by both positive tests with methacholine in 4 of 5 tested patients, and the direct presence of its classic sign - air trapping, in 3 of 4 tested children in the study of external respiration function, possibly caused by hypersecretion characteristic of ARPGA1.
In addition, other important factors that can contribute to inadequate interpretation of the state of the MCC, overestimating it, may be a significant aerodynamic diameter of the radionuclide carrier, which was iron oxide, given its high density (up to 5.24 g / cm3), as well as the lack of consideration of the presence / absence of coughing or mowing, requiring correction for assessing the MCC in case of their presence, despite its prerequisites. Therefore, the understanding of the state of the MCC function remains open, which is fundamental for the pathogenesis of this pathology and requires an adequate study of the MCC in this regard, taking into account these factors and using a standardized radioaerosol method. It is important that this mechanism of reducing the activity of ENC and sodium absorption, on the one hand, plays a pathogenetic role in PGA, on the other hand, indicates the presence of therapeutic potential that can be used in a pathological process in which, on the contrary, there is an increased absorption of sodium and a decrease in periciliary fluid, leading to a violation of the transport of gel and MCC. It has been shown that such a targeted decrease in the activity of ENC, in particular in CF, by epigenetic manipulation due to the inhibition of extracellular peptidases contributes to a decrease in the expression of genes, in particular SCNN 1A, SCNND, SCNN1G, encoding the activity of ENC, and to the restoration of the sol [7,25]. Understanding the pathophysiology of ARPGA1 and its features associated with the MCS can be a good model for improving the understanding of the pathogenesis of CF and similar disorders of ENC in other lung pathologies, including multifactorial ones, opening up new opportunities for optimizing their treatment using epigenetic targeting. As a result, the balance in the mucous link of the MCS is restored, which is so necessary for the normalization of its function and the elimination of exogenous and endogenous material from the DP, eliminating the likelihood of pathogenic microflora development in them by ensuring a balance between the processes of immigration and elimination of opportunistic microorganisms that make up the microbiome of the DP [26]. Candidates for this purpose include camostat,a protease inhibitor; S-Adenosylmethionine (SAM), which has been shown to induce DNA hypermethylation; and curcumin, which is known to induce chromatin condensation. SAM and camostat are drugs that are already clinically used in other pathologies, while curcumin is a common dietary compound [27,28]. As for ARPGA1, it should be emphasized that, given the possibility of implementing only symptomatic therapy for this pathology, a popular new direction in its treatment and conditions similar to it in excess fluid in the DP, for example, distress syndrome, is the development of synthetically obtained cyclic peptides. Their molecular structures, in particular solnatide (TIP, AP301), imitating the lectin-like domain of TNF, contribute to the restoration of mutated ENCs and their transport function [29,30].
Conclusion
Thus, the analysis of the studies conducted on the PGAS model once again demonstrates the significant role of ion transport, and above all sodium transport, in the regulation of the mucous coating and the function of the MCS. In patients with primary systemic PGAS, as a result of tubular sodium reabsorption disorder in the epithelium of the DP, the fluid of the mucous coating is not absorbed in the epithelium of the DP and, as a result, its volume increases. This contributes to a change in the function of the MCS, but the nature of this change is contradictory and requires clarification. An adequate understanding of this will contribute to the improvement of the diagnosis and treatment of patients with disorders in the regulation of the mucous coating of the DP, the function of the MCS, including those caused by disorders in the work of the ENC.
References
- Boucher RC (1994) Human airway ion transport. Am. J. Respir. Crit. Care Med 150: 271-281.
- Stojanoviс V, Spasojeviс S, Radovanoviс T, and Doronjski A (2017) Pseudohypoaldosteronism: report of three cases. Journal of Endocrinology, Metabolism and Diabetes of South Africa 22:17-20.
- Jaudon JC (1948) Further observations concerning hypofunctjon of the adrenal -during early life. J. Pediat 32: 64.
- Hanukoglu A (1991) Type I pseudohypoaldusieronism includes two clinically and genetically distinct entities with either renal or multiple target orcan delects. I Clin Endocrinol Metab 73: 936-944
- Abdalla A, Alhassan MA, Tawfeeg R, Sanad A, Tawamie H, et al (2021) Systemic pseudohypoaldosteronism-1 with episodic dyslipidemia in a Sudanese child. Endocrinol Diabetes Metab Case Rep 2021: 21-0010.
- Chang SS, Grunder S, Hanukoglu A (1996) Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type I. Nat Genet 12: 248-253.
- Strautnieks SS, Thompson RJ, Gardiner RM, Chung E (1996) A novel splice-site mutation in the gamma subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet 13: 248-250
- Aufy M, Hussein AM, Stojanovic T, Studenik CR, Kotob MH (2023) Proteolytic Activation of the Epithelial Sodium Channel (ENaC): Its Mechanisms and Implications. Int J Mol Sci 24:17563.
- Hanukoglu A, Bistriizer T, Rakover Y, Mandelberg A (1994) Pseudohypoal-dosternnism with increased sweat and saliva electrolyte values and frequent lower respiratory tract infections mimicking cystic fibrosisю J. Pediatr 125: 752-755.
- Schaedel C, Marthinsen L, Kristoffersson AC, Kornfält R, Nilsson KO, et al (1999) Lung symptoms in pseudohypoaldosteronism type 1 are associated with deficiency of the alpha-subunit of the epithelial sodium channel. J Pediatr 135: 739-745.
- Silva N, Costa M, Silva A, Sá C, Martins S, et al (2013) A case of systemic pseudohypoaldosteronism with a novel mutation in the SCNN1A gene. Endocrinol Nutr 60: 33-36.
- Hanukoglu A (1991) Type I pseudohypoaldosteronism includes two clinically and genetically distinct entities with either renal or multiple target organ defects. J Clin Endocrinol Metab 73: 936-944.
- Everist J, McVie R (1986) Case report of two children with pseudohypoaldosteronism. Clin Pediatr 25: 44-46.
- Chitayat D, Spirer Z, Ayalon D, Golander A (1985) Pseudohypoaldosteronism in a female infant and her family: Diversity of clinical expression and mode of inheritanceю Acta. Paediatr. Scand 74: 619-622.
- Yager D, Butler JP, Bastacky J (1989) Amplification of airway constriction due to liq-uid filling of airway intersriccs. J. Appl. Physiol 66: 2873-2884.
- Clark JC, Weaver TE, Iwamoto HS, et al (1997) Decreased lung compliance and air trapping in heterozygous SP-B-deficient mice. Am. J. Respir. Cell. Mol. Bio 16: 46-52.
- Hamosh A, Corey M (1993) Cyctic fibrosis genotipe-phenotipe consortium. Corelation between genotipe and phenotipe in patients with cyctic fibrosisю N. Engl. J. Med 329: 1308-1313.
- Amin N, Alvi NS, Barth JH HP, Finlay E, Tyerman K, et al (2013) Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol Diabetes Metab Case Rep 2013:130010.
- Kerem E, Bistritzer T, Hanukoglu A, Hofmann T, Zhou Z, et al (1999) Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N Engl J Med 341:156-162.
- Yoneda K (1976) Mucous blanket of rat bronchus: An ultrastructural study. Am. Rev. Respir. Dis 114: 837-842.
- Yadav E, Yadav N, Hus A, Yadav JS (2020) Aquaporins in lung health and disease: Emerging roles, regulation, and clinical implications. Respir Med 174:106193.
- Kuek LE, Lee RJ (2020) First contact: the role of respiratory cilia in host-pathogen interactions in the airways. Am J Physiol Lung Cell Mol Physiol 319: L603-L619.
- Hessel J, Heldrich J, Fuller J, Staudt MR, Radisch S, et al (2014) Intraflagellar transport gene expression associated with short cilia in smoking and COPD. PLoS One 9: e85453.
- Leopold PL, O'Mahony MJ, Lian XJ, Tilley AE, Harvey BG, et al (2009) Smoking is associated with shortened airway cilia. PLoS One 4: e8157.
- Pugh CP (2022) Pseudohypoaldosteronism Type 1: The Presentation and Management of a Neonate With a Novel Mutation of the SCNN1B Gene Found in Two Hispanic Siblings. Cureus 14: e23918.
- Marrella V, Nicchiotti F, Cassani B (2024) Microbiota and Immunity during Respiratory Infections: Lung and Gut Affair. Int J Mol Sci 25: 4051.
- Martin SL, Saint-Criq V, Hwang TC, Csanády L (2018) Ion channels as targets to treat cystic fibrosis lung disease. J Cyst Fibros 17: S22-S27.
- Blaconà G, Raso R, Castellani S, Pierandrei S, Del Porto P, et al (2022) Downregulation of epithelial sodium channel (ENaC) activity in cystic fibrosis cells by epigenetic targeting. Cell Mol Life Sci 79: 257.
- Buchanan D, Mori S, Chadli A, Panda SS (2025) Natural Cyclic Peptides: Synthetic Strategies and Biomedical Applications. Biomedicines 13: 240.
- Willam A, Aufy M, Tzotzos S, Evanzin H, Chytracek S, et al (2017) Restoration of Epithelial Sodium Channel Function by Synthetic Peptides in Pseudohypoaldosteronism Type 1B Mutants. Front Pharmacol 8: 85.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.