Primary Infratentorial Central Nervous System Neuroblastoma: Case Report and Review from Pediatric Population
by Lisette Alondra Villalobos-Dominguez, Monserrat Perez-Ramirez, Georgina Siordia -Reyes*
Hospital De Pediatría, Centro Médico Nacional Siglo XXI (HPCMNSXXI), Instituto Mexicano Del Seguro Social. Mexico City, Mexico
*Corresponding Author: Georgina Siordia-Reyes, Department of Pathology, Hospital De Pediatría, Centro Médico Nacional Siglo XXI (HPCMNSXXI), Instituto Mexicano Del Seguro Social. Mexico City, Mexico
Received Date: 31 July 2025
Accepted Date: 04 August 2025
Published Date: 06 August 2025
Citation: Alondra LVD, Perez MR, Siordia GR (2025). Primary Infratentorial Central Nervous System Neuroblastoma: Case Report and Review from Pediatric Population. Ann Case Report. 10: 2363. https://doi.org/10.29011/2574-7754.102363
Abstract
Neuroblastomas of the central nervous system are rare malignant neoplasms that predominantly affect pediatric age. The neoplasia can present a wide variety of symptoms depending on its location and size. It has been documented predominantly in the supratentorial and spinal region. The objective is to report an infratentorial pedriatric neuroblastoma case and review of the literature.
Keywords: Central Nervous System; Ganglion Cells; Neurological Deterioration; Neuroblastoma; Hypercellular Tumor.
Introduction
According to the World Health Organization (WHO) 2021 on Central Nervous System (CNS) tumors, neuroblastoma is considered an embryonal neoplasm, characterized by the activity of the transcription factor FOXR2 with chromosomal structural rearrangement [1] gains in chromosome 1q have been found more frequently in about 83-100% of cases [2-4]. Others are in 3q, 8p, 8q, 17q and losses in 3p, 6q, 10q and 16q [5]. Histologically characterized by poorly differentiated cells with round, hyperchromatic nuclei with scant cytoplasm, Homer Wright rosettes and areas of necrosis. Neurocytic differentiation and mature ganglion cells may be present and intense mitotic activity may be present. Immunohistochemistry studies show immunoreactivity to Olig2 and some embryonic cells may show positivity to synaptophysin and Ki67 is usually high [1]. Clinical manifestations present a varied spectrum due to the predominantly supratentorial location of the neoplasm, from focal neurological deficits, irritative signs such as seizures to neurological deterioration [6]. We submit a case of neuroblastoma (NBs) located in the cerebellum and a review of the literature.
Case Presentation
A 9-year-old male, admitted with the complaint of headache, nausea, vomit and ataxia, previously treated as vertigo. After a month and clinical deterioration, a head tomography was performed, and evidence of a cerebellum tumour was found, involving the fourth ventricle with dimensions of 1.2x0.6x0.4 cm in the rostrocaudal, ventrolateral and transverse planes and cerebral edema, for which a ventricle-peritoneal bypass valve was placed. A mid-suboccipital craniotomy was performed with partial resection of 50% of the tumour. He was sent to the HPCMNSXXI for chemotherapy treatment (ifosfamide, carboplatin and etoposide) and radiotherapy. The Pathology service received slides that included routine stains and immunohistochemistry (synaptophysin, GFAP, CD99, Ki67) without paraffin blocks, therefore it was not possible to perform other immunohistochemistry markers or the identification for the transcription factor FOXR2 (forkhead box R2).
An embryonic, hypercellular tumor, consisting of undifferentiated, small and hyperchromatic cells, densely packed with Homer Wright rosettes, which alternated with well-differentiated areas with multiple ganglion cells in different stages of maturation on a fibrillar background was identified. With areas of palisade necrosis and dystrophic calcifications. Immunohistochemistry was positive for synaptophysin in tumor cells, GFAP positive in neuropil, Ki67 positive 95% in neoplastic cells.The diagnosis of Neuroblastoma NOS, grade 4 (WHO 2021), was issued. (Figure 1)
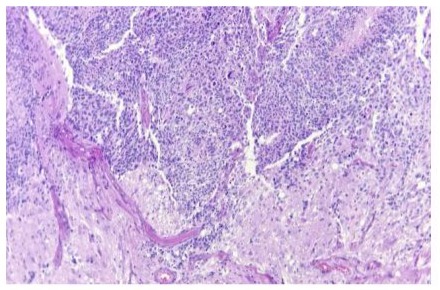
Figure 1: A) HE, Photomicrograph showing a neoplasm consisting of two differentiation components with evidence of embryonic neoplasia alternating with differentiated hypocellular areas 10X
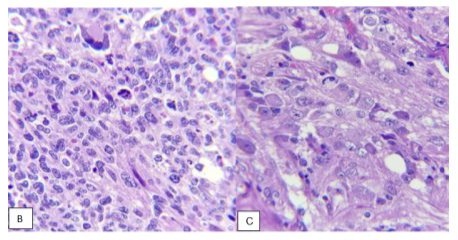
Figure 1: B) Presence of embryonic component with apoptosis and mitosis as well as evidence of multinucleated cell with neuronal differentiation. C) Close-ups in which a fibrillar background is observed with the presence of multiple cells with differentiation neuroblastic or neuronal. (HE, 40x).
Magnetic resonance imaging was performed in April 2023 with the presence of residual lesion in the fourth ventricle that caused a volume effect on the ventricle and adjacent structures (Figure 2). She was followed for 17 months with chemotherapy and radiotherapy treatment, with a torpid evolution followed by death.
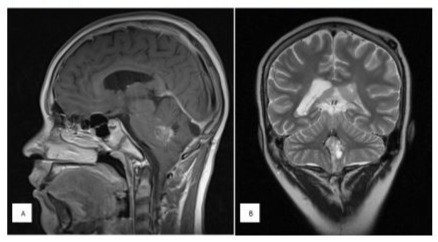
Figure 2: A) Sagittal section in T 1, contrasted with evidence of neoplasm located on the posterior edge of the pons and fourth ventricle that is heterogeneously reinforced. B) Coronal section in T2 showing a hyperintense and heterogeneous midline lesion in the posterior fossa, with few areas with signal intensity similar to CSF.
Material and Methods
A systematic review of published cases in the Web of Science (WOS), SCOPUS, Google Scholar, TripDatabase, Science Direct and PudMed was carried out without using language restrictions, and the search strategy included all known descriptors on CNS neuroblastoma and FOXR2, with no date limit.
The main parameters that were considered eligible for the study were: primary location in CNS and paediatric population.
Other parameters that were included: sex, clinical presentation, surgical treatment, medical treatment, survival time.
The exclusion parameters that were considered for the study were: duplicate cases, CNS metastasis, non-paediatric population, case report, without clinical data of the patients.
The selection process was carried out using Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) 2020, which is summarized in (Figure 3) the studies in this systematic review were carried out between 1966 and 2024, resulting in a final study cohort of 197 patients including our patient.
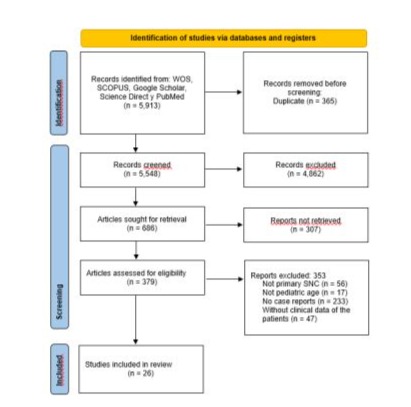
Figure 3: Flow diagram of study identification
Results
(Table 1) shows the summary of the cases found. The criteria were met in 26 publications, plus 6 articles that were not obtained, but the case information was obtained from the publication of Ahdevaara et al., 1976, with a total of 197 cases of NBs from CNS, of which 111 (56.34%) cases were female, and 86 (43.65%) cases were male, with a male:female ratio of 1:1.2.
Only 66 cases (33.5%) documented the most frequent clinical symptoms, those secondary to increased intracranial pressure (headache, nausea, vomiting, convulsions, papilledema), with only ataxia being added in the identified cerebellar case [7].
Of the total number of cases, 195 were supratentorial; 49 frontal, 14 fronto-parietal, 7 fronto-temporal, 2 fronto-temporo-parietal, 9 temporal, 6 temporo-parietal, 3 temporo-parieto-occipital, 23 parietal, 13 parieto-occipital and 6 occipital cases. In the basal nuclei and thalamus 4 cases, intraventricular with 5 cases and 3 cases in other locations. A total of 51 cases were identified as supratentorial lesion, without specifying the affected area. In infratentorial location, a single case was identified located in the right cerebellar lobe and our case, with intraventricular presentation, both cases of the same age at diagnosis and with similar histopathological findings.
Treatment was reported in 92/197 cases (46.7%); 65/92 cases had surgery 33 cases with partial resection and 32 cases with total resection. Chemotherapy was received in 52 cases and 80 cases received radiotherapy. The authors Schmincke 1914*, Horten and Rubinstein 1976, Bennett and Rubinstein 1983, Holsten et al, 2021 and Tietze et al. 2022, with more than 50% (105 patients) of cases included, do not report information on surgical, chemotherapeutic and/or radiotherapy treatment. [8-11]
Of the total number of cases, 62 were reported alive at the time of publication, and the rest were lost and dead as our case. In 102/165 cases, the mean survival was 3 years (0.5 months-14 years), and surgical resection could not be correlated with survival.
|
Author |
No. case |
Sex M/F |
Mean age at diagnosis (min - max) |
Symptoms |
Location |
Resection |
Chemotherapy |
Radiotherapy |
Survival time |
|
Yes / No Partial / Total |
Yes/no |
Yes/no |
Average (min - max) |
||||||
|
Schmincke 1914* |
1 |
M |
17 yr |
ND |
Temporal |
ND |
ND |
ND |
ND |
|
Doyle and Kernohan 1931* |
1 |
F |
11 yr |
ND |
Third ventricle |
Yes |
No |
No |
Alive (30 m) |
|
Kernohan et al., 1932* |
1 |
F |
16 yr |
ND |
Temporo-parietal |
Yes |
No |
No |
Alive (72 m) |
|
Tönnis and Zurich 1939 * |
3 |
M |
3,10, 14 yr |
ND |
Temporo- parietal, parietooccipital 2 |
Yes |
No |
No |
ND |
|
Miller and Ramsden 1966(19) |
1 |
M |
3 m |
Distal cyanosis |
Fronto-parietal |
Yes, total |
No |
No |
Alive (37 m) |
|
Durity et al., 1967(7) |
1 |
F |
3 yr |
Headache, vomiting, hyperreflexia, bilateral positive Babinski and papilledema |
Right cerebellar hemisphere |
Yes, total |
No |
Yes |
Alive (1 yr) |
|
Russell and Rubinstein 1971* |
16 |
8 M |
2.5 m – 9 yr |
ND |
Hemispheres |
Yes |
No |
Yes |
ND |
|
8 F |
|||||||||
|
Henríquez et al., 1973(20) |
1 |
M |
7 yr |
Headache, vomiting and papilledema |
Fronto-parietal |
Biopsy |
Yes |
No |
Dead (15 m) |
|
Rubinstein and Treel 1974* |
1 |
F |
9 yr |
ND |
Parieto-occipital |
Yes |
No |
No |
Alive (24 m) |
|
Horten and Rubinstein 1976(8) |
31 |
17 M |
4 m – 11 yr |
Nausea, vomiting, seizures and headache, |
Frontal 10, parietal 6, temporal 2, fronto-parietal 5, frontotemporal 4, parieto-occipital 2, parietotemporal 1 and parieto-temporooccipital 1 |
ND |
Yes (1 case) |
Yes (8 cases) |
Alive 10 (3m - 13yr) |
|
14 F |
Average 4.1 yr |
Dead 20 (3s -10yr) ND 1 |
|||||||
|
Ahdevaara et al., 1976(16) |
1 |
F |
13 yr |
Headache, vomiting and syncope |
Frontal |
Yes, Partial |
No |
Yes |
Alive (25 m) |
|
Latchaw et al., 1981(21) |
1 (case 1) |
F |
7 yr |
Papilledema |
Nodule in skull |
No |
Yes |
Yes |
Dead (29 m) |
|
Bennett and Rubinstein 1983(9) |
14 M |
5 m - 18 yr |
Increased intracranial pressure 13, seizures 13, hemiparesis 7, headache 2, aphasia 2, macrocephaly 1, hydrocephalus 1 and irritability 1 |
Frontal 14, parietal 4, temporal 2, occipital 2, parieto-occipital 5, fronto-parietal 4, temporoparietal 1, fronto temporo-parietal 2, middle fossa 1, ventricles 2, hemispherical 2 and gyrus rectus 1 |
ND |
ND |
ND |
Alive 21 (18 m-13 yr) |
|
|
40 |
26 F |
Average 5.7 yr |
Dead 12 (0.5 m – 14 yr) ND 7 |
||||||
|
Berger et al., 1983(22) |
10 |
4 M |
17 m – 13 yr |
Headache 4, nausea and vomiting 4, paresthesia of the extremities 4, seizures 2 and decreased memory 2 |
Parietal 3, occipital 3, frontal 2, basal ganglia 1 and temporo-parietal 1 |
Yes, partial 8, total 2 |
Yes (case 3, 4) |
Yes |
Alive 1 (52m) |
|
6 F |
Average 8.2 yr |
Dead 2 (23 and 29 m.) ND 7 |
|||||||
|
Torres et al., 1985(23) |
1 |
F |
3 yr |
Papilledema and syncope |
Temporal |
Yes, partial |
No |
Yes |
Alive (9 years) |
|
PC Davis et al., 1988(24) |
4 |
2 M |
4, 1, 2 a |
Increased intracranial pressure, seizures, vomiting, macrocephaly and ataxia |
Case 6: frontoparietal |
Yes, Partial |
Yes |
Yes |
Alive 3 (28 - 36 m) |
|
(Case 6, 7, 8, 12) |
Case 7: parietal |
Dead 1 (2.5 yr.) |
|||||||
|
Case 8: frontotemporal |
|||||||||
|
Case 12: parieto-temporooccipital |
|||||||||
|
Volkan Etus et al., 2002(25) |
1 |
F |
9:00 AM |
Headache, nausea, papilledema and hemiplegia |
Fronto-parietal |
Yes,Total |
Yes |
Yes |
Alive (8m) |
|
Yarýp et al., 2004(26) |
1 |
F |
5 to |
Headache, vomiting, papilledema and hyperreflexia |
Fronto-parietal |
Yes, partial |
Yes |
Yes |
Alive (34 m) |
|
Ren et al., 2014(27) |
1 |
M |
7:00 AM |
Dizziness, nausea and vomiting |
Thalamus |
Biopsy |
Yes |
Yes |
Alive (16 m) |
|
Bianchi et al., 2018(6) |
1 |
M |
2 to |
Seizures |
Frontal |
Yes, total |
Yes |
No |
Alive (8m) |
|
Mishra et al., 2018(28) |
2 |
1 M |
7 and 12 a.m. |
Headache, vomiting and seizures |
Both lateral ventricles and |
Yes, total |
ND |
ND |
ND |
|
1 F |
Parieto-occipital |
||||||||
|
Furuta et al., 2020(29) |
1 |
F |
3 to |
Anorexia and vomiting |
Frontal |
Yes, partial |
Yes |
Yes |
Alive (24 m) |
|
Lastowsky et al., 2020(15) |
3 |
2 M |
5, 4.5, 7 to |
ND |
Parietal, parietooccipital, frontal |
Yes, total |
Yes |
Yeah |
ND |
|
1 F |
|||||||||
|
Holsten et al., 2021(10) |
8 |
4 M |
1 to – 9 to |
ND |
Frontal 3, occipital 1, parieto-occipital 1, parietotemporooccipital 1, frontotemporal 1 and intraventricular 1 |
ND |
ND |
ND |
Alive 4 (139, 12, 31, 8 m.) |
|
4 F |
Average 5 to |
Dead 2 (18, 26 m) |
|||||||
|
ND 2 |
|||||||||
|
Borni et al., 2021(18) |
1 |
M |
6:00 AM |
Headache, vomiting and papilledema |
Fronto-temporal |
Yes, total |
No |
No |
ND |
|
Taschner et al., 2021(30) |
1 |
F |
6:00 AM |
Vomiting and facial paralysis |
Temporo-parietal |
Yes, partial |
Yes |
No |
Alive (4 s) |
|
Korshunov et al., 2021(4) |
20 |
5 M |
4 a – 16 a |
ND |
Hemispherical |
Yes, partial 10, total 10 |
Yes |
Yes |
ND |
|
15 F |
Average 8 to |
||||||||
|
Shimazaki et al., 2022(17) |
3 |
1 M |
6.7, 1.6, 2.6 a |
Nausea, vomiting and convulsions |
Frontal 2, |
Yes |
Yes (case 1) |
Yes (case 1) |
Alive (7, 66, 1 m) |
|
2 F |
Parietal 1 |
||||||||
|
Tietze et al., 2022(11) |
25 |
12 M 13 F |
1.4 a – 16 a |
A case with seizures |
Frontal 13 |
ND |
ND |
ND |
ND |
|
Average 4.5 to |
Parietal 7 |
||||||||
|
Temporal 3 |
|||||||||
|
Basal ganglia 2 |
|||||||||
|
Liu et al., 2022(3) |
6 |
3 M |
0.91 a - 11.9 a |
ND |
Hemispheres |
Yes, partial 2, total 5 |
Yes |
Yes (4 cases) |
ND |
|
3 F |
Average: 1.7 a |
||||||||
|
Schepke et al., 2023(2) |
7 |
3 M |
3 a – 15.7 a |
ND |
Hemispherical |
Yes, partial 2, total 5 |
Yes |
Yeah |
Alive (5 to) |
|
4 F |
Average 5.3 a |
||||||||
|
Sharkey et al., 2024(31) |
1 |
M |
2 to |
Seizures |
Frontal |
Yes, partial |
Yes |
Yeah |
ND |
|
Villalobos and Siordia 2024 |
1 |
M |
9:00 AM |
Headache, nausea, vomiting and ataxic gait |
Cerebellum |
Yes, partial |
Yes |
Yes |
Dead (17m) |
Table 1: Literature review of reported cases of primary CNS neuroblastoma
M: male F: female ND: not available a: years m: months s: weeks
*The papers were not gotten and the cases. Were referenced in Ahdevaara et al., 1976.
Discussion
CNS neuroblastoma, FOXR2-activated, is a rare, recently described tumor that presents in children. They correspond to 10% of all neoplasms previously referred to as primitive neuroectodermal tumors of the CNS. [1] There are cases of FOXR2 Neuroblastoma in the literature, diagnosed as ependymomas or glioblastomas prior to the WHO 2021 classification. [12]
CNS neuroblastoma originates from neuroectodermal cells, although the exact cell that gives rise to it is not clear. Histologically, it is characterized by a poorly differentiated pattern, many of them being previously diagnosed as primitive neuroectodermal tumor of the CNS due to the embryonic characteristics constituted by small, round hyperchromatic nucleus cells with scant cytoplasm, alternating with areas with clear cells with the appearance of neurocytic differentiation. The differentiated areas show ganglion cells on a histologically fibrillar background like a ganglioneuroblastoma, the presence of both differentiation patterns is suggestive of CNS FOXR2 NBs similar histological appearance that is observed in the present case. Necrosis, high mitotic index, the presence of Homer-Wright rosettes, as well as infiltration into adjacent parenchyma are evident as in any other embryonal tumor. Immunohistochemical markers [1,10] are nonspecific, usually positive for Olig2, synaptophysin, NeuN, MAP2C, SOX10 and 1q gain by FISH with sensitivity and specificity of 100% [4,10,13]. Fibrillar glial acid protein staining is occasionally identified, although there are publications that report that it is not expressed in this type of tumor. The Ki67 proliferation index is high. Holsten et al., reported in eight cases, range from 12 to 50% with an average of 26% positive cells [1,10] FOXR2 is a transcription factor located on the X chromosome and its activation is associated with hypomethylation. The expression is absent in almost all normal postnatal human tissues and has been related as an oncogene in a large number of neoplasms of different types of cancer that represent 71% of the most frequent neoplasms (melanoma, endometrial cancer, non-small cell lung cancer) in adults and in pediatrics in neuroblastoma, sarcomas and diffuse midline glioma with different epigenetic and genetic mechanisms of their expression. In neuroblastomas, the detection of FOXR2 has been studied only in supratentorial tumors, and they have not been described in the infratentorial region, which can be expected due to the rarity of the presentation. To date, 70 cases of the entire review had FOXR2 detected. [10,11,14,15]
In the literature, it is reported that 85% of cases of CNS neuroblastoma occur in the first decade and 65% in the first 5 years of age. The mean age was 5.5 years with a range of 2 weeks to 18 years of age. [16] The clinical manifestations are nonspecific, although due to their mainly supratentorial presentation, signs associated with intracranial hypertension (vomiting and headache), neurocognitive impairment and seizures predominate; in infants the signs and symptoms are not so evident, due to the fact that the sutures have not fused. The two cases of infratentorial presentation were characterized by cerebellar signs such as ataxic gait. [6]
Dissemination at diagnosis has only been reported in the literature in two cases of CNS NBs; one of them to the neuroaxis by seeding through the CSF and extracranial metastasis (lymph node) in another case, in the present case they were not documented. [6]
Radiographically, CNS NBs are characterized by being heterogeneous with solid and cystic components or pure solids, and the findings of tomography (T1 and T2) and magnetic resonance imaging are nonspecific for this entity. In a systematic review conducted by Shimazaki et al. in 34 cases, the frontal lobe was reported to be more frequent in 67.6%, cortical involvement in
85.3%, deep white matter in 97.1%, and in the ventricular system in 61.8%. Spectroscopy is expected to indicate the accentuated choline peak indicating synthesis and degradation of the membrane being an indicator of hypercellularity [11,17] . A finding documented in the literature and observed in our experience is the fact that embryonal tumors of the CNS, including in NBs, are characterized by producing little or no peritumoral edema [10,11,17].
The survival reported by Von Hoff et al. 2021 mentions that the 5-year progression-free survival (PFS) is 63% (5) + 8% and the 5-year overall survival (OS) of 85% + 5%, is very similar with Liu et al. 2022 with 5-year PFS of 66.7% (3) + 19.2% and 5-year OS of 83.3% + 15.2%, on the other hand, Schepke et al. 2023 report that in their cases PFS and OS at 5 years was 100% in both and refers to the fact that all their patients received treatment with high-dose radiotherapy in the CNS and neuraxis, which for them was the factor that favored their patients [2].
Surgical resection is one of the prognostic factors of great importance for the survival of CNS neoplasms, and treatment is based on American, European or Japanese protocols, in which they all have the same chemo agents, but with different application schemes. Although radiation therapy is the treatment of choice, its usefulness is limited in children younger than 3 years of age. The review that was conducted did not specify survival related to the type of resection (partial or total) in most of the cases reported. The present case survived 17 months to a partial resection of 50% coupled with chemotherapy and radiotherapy, well below than previously reported. [18-31]
References
- Louis DN, Perry A, Wesseling P (2021) Central nervous system tumours. International Agency for Research on Cancer. 5: 232-234.
- Schepke E, Löfgren M, Pietsch T, Kling T, Nordborg C, et al (2023) Supratentorial CNS-PNETs in children; a Swedish population-based study with molecular re-evaluation and long-term follow-up. Clin Epigenetics. 15.
- Liu APY, Dhanda SK, Lin T, Sioson E, Vasilyeva A, et al (2022) Molecular classification and outcome of children with rare CNS embryonal tumors: results from St. Jude Children’s Research Hospital including the multi-center SJYC07 and SJMB03 clinical trials. Acta Neuropathol. 144: 733-746.
- Korshunov A, Okonechnikov K, Schmitt-Hoffner F, Ryzhova M, Sahm F, et al (2021) Molecular analysis of pediatric CNS-PNET revealed nosologic heterogeneity and potent diagnostic markers for CNS neuroblastoma with FOXR2-activation. Acta Neuropathol Commun. 9: 20.
- Von Hoff K, Haberler C, Schmitt-Hoffner F, Schepke E, De Rojas T, et al (2021) Therapeutic implications of improved molecular diagnostics for rare CNS embryonal tumor entities: Results of an international, retrospective study. Neuro Oncol. 23: 1597-611.
- Bianchi F, Tamburrini G, Gessi M, Frassanito P, Massimi L, et al (2018) Central nervous system (CNS) neuroblastoma. A case-based update. Child’s Nervous System. 34: 817-823.
- Durity FA, Dolman CL, Moyes PD (1967) Ganglioneuroblastoma of the Cerebellum Case Report. Vancouver. 28: 270-3.
- Horten BC, Rubinstein LJ (1976) Primary cerebral neuroblastoma: A clinicopathological study of 35 cases. Brain. 99: 735-56.
- Bennett JP, Rubinstein LJ (1984) The biological behavior of primary cerebral neuroblastoma: A reappraisal of the clinical course in a series of 70 cases. 16: 21-7.
- Holsten T, Lubieniecki F, Spohn M, Mynarek M, Bison B, et al (2021) Detailed clinical and histopathological description of 8 cases of molecularly defined CNS neuroblastomas. J Neuropathol Exp Neurol. 80: 52-59.
- Tietze A, Mankad K, Lequin MH, Ivarsson L, Mirsky D, et al (2022) Imaging characteristics of CNS neuroblastoma-FOXR2: A retrospective and multi-institutional description of 25 cases. American Journal of Neuroradiology. 43: 1476-1480.
- Sturm D, Orr BA, Toprak UH, Hovestadt V, et al (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 164: 1060-1072.
- Tauziède-Espariat A, Figarella-Branger D, Métais A, Uro-Coste E, Maurage CA, et al (2023) CNS neuroblastoma, FOXR2-activated and its mimics: a relevant panel approach for work-up and accurate diagnosis of this rare neoplasm. Acta Neuropathol Commun. 11: 43
- Tsai JW, Cejas P, Wang DK, Patel S, Wu DW, et al (2022) FOXR2 is an epigenetically regulated pan-cancer oncogene that activates ETS transcriptional circuits. Cancer Res. 82: 2980-3001.
- Lastowska M, Trubicka J, Sobocińska A, Wojtas B, Niemira M, et al (2020) Molecular identification of CNS NB-FOXR2, CNS EFT-CIC, CNS HGNET-MN1 and CNS HGNET-BCOR pediatric brain tumors using tumor-specific signature genes. Acta Neuropathol Commun. 8: 105.
- Ahdevaara P, Kalimo H, Haltia M (1977) Differentiating intracerebral neuroblastoma: Report of a case and review of the literature. 40: 7848.
- Shimazaki K, Kurokawa R, Franson A, Kurokawa M, Baba A, et al (2023) Neuroimaging features of FOXR2-activated CNS neuroblastoma: A case series and systematic review. Journal of Neuroimaging. 33: 359367.
- Borni M, Znazen M, Mdhaffar N, Boudawara MZ (2021) A rare case of pediatric primary central nervous system differentiating neuroblastoma: An unusual and rare intracranial primitive neuroectodermal tumor (a case report). Pan African Medical Journal. 40: 33.
- Miller AA, Ramsden F (1966) A cerebral neuroblastoma with unusual fibrous tissue reaction.
- Henriquez AS, Robertson DM, John W, Marshall S (1973) Primary neuroblastoma of the central nervous system with spontaneous extracranial metastases: Case report. 38: 226-231.
- Latchaw RE, L’heureux PR, Young G, Priests JR (1981) Neuroblastoma presenting as central nervous system disease. 3: 623-30.
- Berger MS, Edwards MSB, Wara WM, Levin VA, Wilson CB (1983) Primary cerebral neuroblastoma: Long-term follow-up review and therapeutic guidelines. J Neurosurg. 59: 418-23.
- Torres LF, Grant N, Harding BN, Scaravilli F (1985) Intracerebral neuroblastoma: Report of a case with neuronal maturation and long survival. Acta Neuropathol (Berl). 68: 110-4.
- Davis PC, Wichman RD, Takei Y, Hoffman JC, All A (1990) Primary cerebral neuroblastoma: CT and MR findings in 12 cases. American Society of Neuroradiology. 11: 115-120.
- Etus V, Kurtkaya Ö, Sav A, Ilbay K, Ceylan S (2002) Primary cerebral neuroblastoma: A case report and review. J Exp Med. 197: 55-65.
- Yariş N, Yavuz MN, Reis A, Yavuz A, Ökten A (2004) Primary cerebral neuroblastoma: A case treated with adjuvant chemotherapy and radiotherapy. The Turkish Journal of Pediatrics. 46: 182-5.
- Ren AJ, Ning HY, Lin W (2014) Serial diffusion-weighted and conventional MR imaging in primary cerebral neuroblastoma treated with radiotherapy and chemotherapy: A case report and literature review. Neuroradiology Journal. 27: 417-421.
- Mishra A, Beniwal M, Nandeesh BN, Srinivas D, Somanna S (2018) Primary pediatric intracranial neuroblastoma: A report of two cases. J Pediatr Neurosci. 13: 366-370.
- Furuta T, Moritsubo M, Muta H, Koga M, Komaki S, et al (2020) Central nervous system neuroblastic tumor with FOXR2 activation presenting both neuronal and glial differentiation: A case report. Brain Tumor Pathol. 37: 100-104.
- Taschner U, Diebold M, Shah MJ, Prinz M, Urbach H, et al (2021) Freiburg Neuropathology Case Conference: A 6-year-old girl presenting with vomiting and right-sided facial paresis. Clin Neuroradiol. 31: 885892.
- Sharkey B, Conner KM, McGarvey CR, Nair A, Dorn A, et al (2024) Pediatric central nervous system (CNS) neuroblastoma: A case report. Surg Neurol Int. 15: 1-5.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.