Pretibial Dystrophic Epidermolysis Bullosa Successfully Treated with Upadacitinib: A Case Report
by Romina Caushaj, Francesco Bellinato*, Paolo Gisondi, Giampiero Girolomoni
Section of Dermatology and Venereology, Department of Medicine, University of Verona, Verona, Italy
*Corresponding author: Francesco Bellinato. Section of Dermatology and Venereology, Department of Medicine, University of Verona, Piazzale A. Stefani 1, 37126 Verona, Italy
Received Date: 24 August 2024
Accepted Date: 28 August 2024
Published Date: 30 August 2024
Citation: Caushaj R, Bellinato F, Gisondi P, Girolomoni G (2024) Pretibial Dystrophic Epidermolysis Bullosa Successfully Treated with Upadacitinib: A Case Report. Ann Case Report. 9: 1951. https://doi.org/10.29011/2574-7754.101951
Abstract
Pretibial dystrophic epidermolysis bullosa (PDEB) is a rare subtype of dystrophic epidermolysis bullosa caused by mutations in the gene encoding type VII collagen, and is characterized by intensely itchy papular, plaque and bullous lesions on the shin area. Herein, we report a 58-year-old man with recessive PDEB, successfully treated with upadacitinib 30 mg daily, a selective JAK1 inhibitor. The patient presented multiple tense bullae, purplish-red papular-nodular lesions, and severe pruritus (VAS 10/10). Previous treatments, including clobetasol and tacrolimus cream, prednisone, cyclosporine and dupilumab, were only minimally effective. After one month, significant clinical improvement was observed with minimal lesions and substantial pruritus reduction (VAS 0/10). No drug-related side effects or laboratory abnormalities were noticed. Upon drug discontinuation, the disease recurred in few days; nevertheless reintroducing upadacitinib 30 mg daily resulted in similar clinical benefits. Further studies are needed to assess whether JAK inhibitors may offer effective symptomatic relief for PDEB patients.
Keywords: Pretibial dystrophic epidermolysis bullosa; JAKInhibitors; Upadacitinib; refractory epidermolysis bullosa.
Introduction
Pretibial dystrophic epidermolysis bullosa (PDEB) is a rare subtype of dystrophic epidermolysis bullosa, a group of inherited skin disorders characterized by blister formation and skin fragility due to mutations in the gene encoding type VII collagen, a crucial component of the anchoring fibrils of the cutaneous basement membrane zone (BMZ) [1].
PDEB is characterized by the formation of itchy blisters, milia, atrophic scarring, and lichenoid papules and plaques localized in the pretibial region and it can be associated with dystrophic or absent toenails [2,3]. Disease onset is generally during early childhood, but sometimes it can be delayed until the second or third decades [3]. The prevalence of PDEB is estimated to be less than 1 in 1,000,000; however, it might be underestimated due to the frequent misdiagnoses, which often result in delayed treatments [4]. Localized bullous and lichenoid dermatoses, as well as eczema and prurigo nodularis may mimic PDEB [5,6]. Since there is currently no targeted therapy for PDEB, patient management is primarily supportive, focusing on relief from itching, dampening inflammation, wound care and the prevention of complications [7].
Case Presentation
We report the case of a 58-year-old man with PDEB, successfully treated with upadacitinib. The disease began during childhood, with progressive clinical deterioration over the second and third decades. None of the family members were affected by the same pathological condition. Physical examination showed multiple tense bullae and purplish-red papular nodular lesions on the legs, with widespread hemorrhagic crusts and superimposed scratching lesions, as well as toenail dystrophy (Figure 1A). The patient reported severe pruritus, significantly impairing his sleep and daily activities (pruritus visual analogue scale (VAS) 10/10, Dermatology Life quality index (DLQI) 15/30). Histopathology revealed the presence of subepidermal blisters with a cleavage plane beneath the lamina densa of the basement membrane and a dense band of inflammatory cells in the upper dermis, predominantly composed of lymphocytes. Direct immunofluorescence (DIF) mapping showed linear fluorescence for collagen IV (mAb CIV22) along the dermoepidermal and dermo-adnexal junction above the cleavage plane. Transmission electron microscopy (TEM) showed a numerical reduction and hypoplastic appearance of the anchoring fibrils beneath the lamina densa (Figure 2). Molecular investigations confirmed a compound heterozygosity due to c.3355C>T and c.5820G>A mutations in the COL7A1 gene. The patient’s medical history was unremarkable for other diseases.
Previous treatments included topical clobetasol propionate 0.05% cream, topical tacrolimus 0.1% ointment and oral prednisone 25 mg daily, which provided partial relief. Cyclosporine 300 mg daily led to temporary improvement but relapse upon discontinuation.
Subsequent therapy with dupilumab 300 mg every two weeks for four months yielded no benefits.
The patient was then prescribed upadacitinib 30 mg daily. After one month, significant clinical improvement was noted, with marked reduction of papular and nodular lesions, absence of scratching lesions and hemorrhagic crusts (Figure 1B). Healed areas displayed multiple milia. The patient reported a substantial reduction in pruritus and enhanced quality of life (pruritus VAS 0/10, DLQI 2/30).
During the 6-month follow-up period, the patient did not experience any drug-related side effects or abnormalities in routine laboratory tests. Upon treatment discontinuation, given the minimal disease activity, a recurrence of the disease was observed, along with an increase in pruritic symptoms and deterioration in quality of life (VAS 8/10, DLQI 10/30). Consequently, upadacitinib was reintroduced at the original dosing regimen, achieving complete clinical remission.
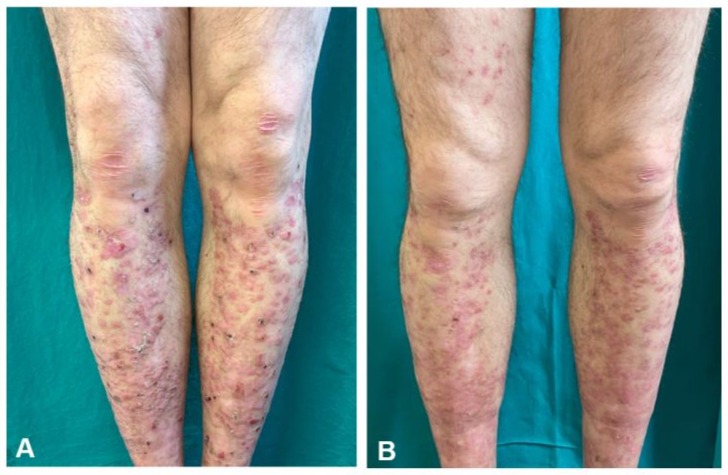
Figure 1: Papules, plaques, and crusts on both pretibial areas, along with a few small vesicles, milia, and atrophic erythematous scars with erosions attributable to scratching. (A) Following one month of upadacitinib treatment, the erosions and blisters have disappeared. (B).
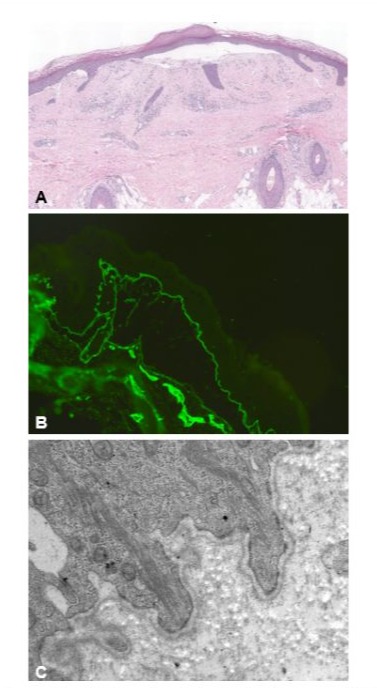
Figure 2: Histopathology showing subepidermal bulla formation and a dense band of inflammatory cells in the upper dermis, predominantly composed of lymphocytes (haematoxylin & eosin, 4x) (A). Direct immunofluorescence showing deposits of collagen IV located on the epidermal side of the separation area (B). Transmission electron microscopy revealing basal keratinocytes, hemidesmosomes, regular insertion of tonofilaments, normal appearance of lamina rara and lamina densa, and numerical reduction and hypoplastic appearance of the anchoring fibrils at the deep side of the lamina densa (C).
Discussion
PDEB is a rare condition with no currently available treatment . Management involves supportive care, which includes wound care, infection control, and prevention or early treatment of complications. Topical or systemic corticosteroids and topical immunomodulators are considered appropriate first-line therapies. Additionally, narrow-band UVB phototherapy, etretinate, cyclosporine, thalidomide, and naltrexone have also been documented as therapeutic options [8]. Refractory pruritus is a particularly distressing symptom for patients with EB, perpetuating a vicious itch-vesicle cycle, leading to further skin damage and discomfort [9]. Despite the uncertainty regarding the specific key itch mediators, the involvement of Th2-mediated inflammation in PDEB has been long suspected [9,10]. Mechanical trauma can trigger an inflammatory response with a Th2 profile at the injury site, involving T cells, eosinophils, macrophages, and mast cells. This response releases itch mediators like IL-4, IL-13, and IL31, which may explain why dupilumab, an IL-4 alpha receptor inhibitor, is effective in some cases [11,12]. Additionally, the JAK/ STAT pathway plays a crucial role in transmitting Th2 cytokine signals in PDEB, and JAK inhibitors have shown efficacy in treating this condition [13,14,15].
Our case highlights the potential of JAK1 inhibition in providing significant symptomatic relief for patients with PDEB, particularly in reducing refractory pruritus, which is a major cause of discomfort and further skin damage. Further studies are needed to confirm the efficacy and safety of JAK inhibitors in larger series of PDEB patients.
Acknowledgment: No Acknowledgments.
Informed Consent Statement: The authors obtained written consent from patients for their photographs and medical information to be published in print and online and with the understanding that this information may be publicly available. Patient consent forms were not provided to the journal but are retained by the authors.
Data Availability Statement: the data used and analysed in this study are available from the corresponding author on reasonable request.
Funding statement: This article has no funding source.
Conflict of interest statement: The Authors have no conflict of interest to declare. The Authors do not hold any stock or shares in any entity that may gain financial benefit or detriment as a result of the deliberations set out in or the conclusions of the study.
References
- Has C, Bauer JW, Bodemer C, Bolling MC, Bruckner-Tuderman L, et al. (2020) Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 183:614627.
- Callegaro EAC, Nappi F, Lazzarini R, Lellis RF. (2017) Pretibial dystrophic epidermolysis bullosa. An Bras Dermatol. 92:126-128.
- Lee YY-J, Chen H-C, Lin S-J. (1993) Pretibial epidermolysis bullosa: a clinicopathologic study. J Am Acad Dermatol. 29:974-981.
- Tang WY, Lee KC, Chow TC, Lo KK. (1999) Three Hong Kong Chinese cases of pretibial epidermolysis bullosa: a genodermatosis that can masquerade as an acquired inflammatory disease. Clin Exp Dermatol. 24:149-153.
- Bridges AG, Mutasim DF. (1999) Pretibial dystrophic epidermolysis bullosa. Cutis. 63:329-332.
- Cunningham L, Liu L, Menzies S, McGrath JA, Lally A. (2015) Novel missense mutation in a patient with recessive pretibial epidermolysis bullosa and a mild phenotype. J Eur Acad Dermatol Venereol. 30: e115-e116.
- Pabón-Carrasco M, Caceres-Matos R, Roche-Campos M, HurtadoGuapo MA, Ortiz-Romero M, et al. (2024) Management of Skin Lesions in Patients with Epidermolysis Bullosa by Topical Treatment: Systematic Review and Meta-Analysis. Healthcare (Basel). 12:261.
- Vaccaro M, Guarneri C, Guarneri F, Lentini M, Cannavò SP. (2020) Dominant pretibial dystrophic epidermolysis bullosa in an Italian family. Pediatr Dermatol. 37: 1207-1209.
- Papanikolaou M, Nattkemper L, Benzian-Olsson N, Liu L, Guy A, et al. (2024) Th2 response drives itch in dystrophic epidermolysis bullosa pruriginosa: a case-control study. J Am Acad Dermatol. 91: 130-133.
- Caroppo F, Milan E, Giulioni E, Belloni Fortina A. (2022) A case of dystrophic epidermolysis bullosa pruriginosa treated with dupilumab. J Eur Acad Dermatol Venereol. 36: e365-e367.
- Bellon N, Bataille P, Bonigen J, Charbit-Henrion F, Dietrich C, et al. (2024) Experience of dupilumab treatment in inherited epidermolysis bullosa: A short series. J Am Acad Dermatol. 91: 373-376.
- Clawson RC, Duran SF, Pariser RJ. (2021) Epidermolysis bullosa pruriginosa responding to dupilumab. JAAD Case Rep. 16:69-71.
- Damsky W, King BA. (2017) JAK inhibitors in dermatology: The promise of a new drug class. J Am Acad Dermatol. 76:736-744.
- Kwon IJ, Kim SE, Kim SC, Lee SE. (2024) Efficacy of oral JAK1 or JAK1/2 inhibitor for treating refractory pruritus in dystrophic epidermolysis bullosa: A retrospective case series. J Dermatol. 51:441-447.
- Zhang Z, Lin Z. (2024) Epidermolysis bullosa pruriginosa treated with upadacitinib. JAMA Dermatol. 21: e242787.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.