Multidisciplinary Real-World Evaluation of Switching to the Oral Chaperone Therapy Migalastat in Fabry Disease with the S238N Mutation Short Running Title: Switching to Migalastat in Fabry Disease
by Raúl Noguera1,2, Rosario Sánchez Martínez2,3,4, Elena García-Payá4,5, Vicente Climent-Payá4,6*
1Nephrology Department, Hospital General Universitario Dr. Balmis de Alicante, Alicante, Spain
2Multidisciplinary Unit for Low Prevalence Diseases, Hospital General Universitario Dr. Balmis de Alicante, Alicante, Spain
3Internal Medicine Department, Hospital General Universitario Dr. Balmis de Alicante, Alicante, Spain
4Institute for Health and Biomedical Research (ISABIAL), Alicante, Spain
5Laboratory of Molecular Cytogenetics, Hospital General Universitario de Dr. Balmis de Alicante, Alicante, Spain
6Inherited Cardiovascular Diseases and Cardiac Imaging Unit. Department of Cardiology. Hospital General Universitario Dr. Balmis de Alicante, Alicante, Spain
*Corresponding author: Vicente Climent-Payá, Institute for Health and Biomedical Research (ISABIAL), Alicante, Spain; Inherited Cardiovascular Diseases and Cardiac Imaging Unit. Department of Cardiology. Hospital General Universitario Dr. Balmis de Alicante, Alicante, Spain
Received Date: 25 November 2024
Accepted Date: 29 November 2024
Published Date: 02 December 2024
Citation: Noguera R, Martinez RS, Garcia-Paya E, Climent-Paya V (2024) Multidisciplinary Real-World Evaluation of Switching to the Oral Chaperone Therapy Migalastat in Fabry Disease with the S238N Mutation Short Running Title: Switching to Migalastat in Fabry Disease. Ann Case Report. 9: 2097. https://doi.org/10.29011/2574-7754.102097
Abstract
Introduction: Spain has an increased prevalence of Fabry Disease (FD) caused by the S238N mutation in the GLA gene. Currently, there is no real-world data on the management of these patients. Here, we report the course of an S238N FD cohort that switched from enzyme replacement therapy (ERT) to migalastat.
Material and Methods: Adult patients with FD and carrying the S238N mutation who were treated with ERT for >18 months followed by migalastat treatment for >2 years were evaluated. We performed a multidisciplinary assessment of the effect of the switch from ERT to migalastat using information from the patient’s medical charts.
Results: Seven men were followed up for >9 years. All patients with available data had decreased α-Gal A activity in white blood cells and increased plasma Lyso-Gb3 levels at the time of the switch, as well as advanced cardiac disease. Enzyme activity of all patients increased or was maintained after 24 months of migalastat treatment. Renal and cardiac function parameters were maintained during the migalastat treatment. No cardiological clinical events were reported.
Conclusions: Renal and cardiac function parameters remained stable after switching from ERT to migalastat in FD patients exhibiting the S238N mutation. The treatment switch was well tolerated and safe.
Keywords: Fabry Disease; alpha-Galactosidase; Inborn Genetic Diseases, X-Linked Genetic Diseases
Introduction
Fabry Disease (FD) is an X-linked lysosomal storage disorder with multiple organ involvement. A deficiency in the enzyme alphagalactosidase A (α-Gal A) results in intracellular accumulation of glycosphingolipids. Progressive fibrosis causes neurological dysfunction, myocardial hypertrophy, and heart and kidney failure. In the end, FD patients require dialysis and organ transplants. Preventing irreversible organ damage requires timely diagnosis and treatment. In addition to supportive care, migalastat (for patients with amenable gene mutations i.e., 35%-50% of patients) and enzyme replacement therapy (ERT) are currently available for FD [1]. FD is a rare disease with recent screening studies revealing higher frequencies than previously estimated. These studies, which focused on male newborns, found incidence rates ranging from 1:316 to 1:7575 [2,3]. In some geographical areas, the presence of founder mutations has further increased the local prevalence of FD. The S238N mutation previously described in isolated cases [4], was later found to be the most prevalent mutation in Spanish females with FD, constituting 31% of cases [5]. One study, conducted in Alicante’s Elda Health Department involving 42 patients with the S238N mutation, confirmed its high prevalence in the region [6]. S238N (NM_000169.3:c.713G>A; p.Ser238Asn; GRCh38) is a missense mutation in exon 5 of the GLA gene that substitutes Serine for Asparagine at position 238 of α-Gal A [4]. FD caused by the S238N mutation has been described as a later onset variant affecting the heart and kidneys [4,6].
Fabry patients with this specific mutation are prevalent in Spain, particularly Alicante. Considering the non-standardized nature of follow-up of this disease, as well as the lack of real-world data relating to switching from ERT to migalastat [7,8], a more extensive analysis of this population may be necessary. Therefore, in this study we describe the results of the multidisciplinary evaluation in seven FD patients who share the same mutation and their outcomes after switching to migalastat.
Materials and Methods
This is a descriptive case series of seven male patients with FD from the Hospital General Universitario Dr. Balmis de Alicante (Alicante, Spain), aged ≥18 years and carrying the S238N mutation. The patients had all been treated with ERT (agalsidase alfa) for at least 18 months and subsequently with migalastat for at least two years. We evaluated the outcomes of switching from ERT to migalastat at baseline and at 6, 12 and 24 months after starting migalastat. Information was collected from patients’ medical charts, including demographics, personal medical histories, comprehensive organ-specific investigations, and laboratory and instrumental exams to assess the organ involvement in FD patients.
As part of clinical practice in our hospital, the following methodology was used: α-Gal A enzyme activity and plasma levels of Lyso-Gb3 were measured in dried blood spots; until 2018, renal function was measured by changes in urine albumin-creatinine ratio, and by eGFR quantified by the Modification of Diet in Renal Disease-4 [MDRD4] equation [9], and then by the Chronic Kidney Disease Epidemiology Collaboration [CKD-EPI] formula [10]); a combination of echocardiography (thickness of cardiac structures, left ventricular volume, measures of systolic and diastolic function, heart rate) and electrocardiography, as well as plasma troponin T and pro–brain natriuretic peptide (NT-proBNP) concentration were used to assess cardiac changes.
Results
The characteristics of patients are described in Table 1. Seven men, three of whom were unrelated, plus two sets of brothers, all from five unrelated families, were assessed with a mean (range) of follow-up (pre- and post-switch) of 9.2 (4.3-14.9) years. Four patients switched to migalastat due to progression or lack of cardiologic response, two due to the patient’s own choice, and one due to poor venous access. Clinical and biochemical findings at ERT and migalastat start are described in (Table 2). All patients, except one who did not have the measurement at migalastat start, had decreased α-Gal A activity in white blood cells with levels below 80% of the reference value (≥15.3 μmol/L/h), with three patients with almost no residual activity (patients 2, 3 and 6), and increased plasma Lyso-Gb3 levels, reference value: ≤1.8 ng/ mL). The eGFR prior to the commencement of migalastat was normal (>90 mL/min/1.73 m2) in five patients, and there were two patients with baseline CKD stages 2 (60-89 mL/min/1.73 m2) and 3 (30-59 mL/min/1.73 m2). Six out of seven patients registered microalbuminuria (30-300 mg/g) before migalastat was initiated, one of them had undergone a kidney transplant. Cardiac magnetic resonance imaging revealed increased thickness of the interventricular septum (IVS) (normal ≤12 mm) and left ventricular hypertrophy (LVH) in four patients, with a left ventricular mass index (LVMi) ranging from 142 to 260 g/m2 (normal: 115 g/m2). In four patients, an early transmitral velocity/tissue Doppler early diastolic mitral annular velocity (E/Ea) >15 indicated increased pulmonary capillary pressure (PCP), reflecting the left atrial pressure or elevation in left ventricular filling pressure. Elevated cardiac troponin T and NT-proBNP (normal range: 0-14 ng/L and 0-150 pg/mL, respectively) indicated advanced heart disease in three.
In all patients, enzyme activity increased 6-12 months after migalastat start, and the achieved levels of enzyme activity were maintained with the exception of one patient, who returned to almost residual baseline levels at month 24 with an increase in plasma Lyso-Gb3 (Figure 1).
After 24 months of treatment with migalastat, the stage of CKD remained the same in all the patients. Microalbuminuria values also remained stable during the treatment period, with a slight reduction in most patients (Figure 1). The patient with proteinuria presented a slight reduction in the levels from baseline (1279 mg/g creatinine [Cr]) to 24 months after treatment with migalastat (1104 mg/g Cr). In the case of the kidney-transplanted patient, renal parameters remained stable, and no adjustments to immunosuppressive treatments were necessary.
All patients showed stable LVH and cardiac function during treatment with migalastat (Figure 1). In four patients, the diastolic function marker E/Ea decreased and, specifically, in patient 7, whose baseline E/Ea was 17.8, at month 24 it was below 14, indicating an improvement of diastolic function with lowered LV filling pressure than before treatment with migalastat. There were only two cases in which E/Ea increased; one patient remained stable (Figure 1). Treatment with migalastat was generally safe and well tolerated. Patients did not experience any cardiological clinical events during follow-up.
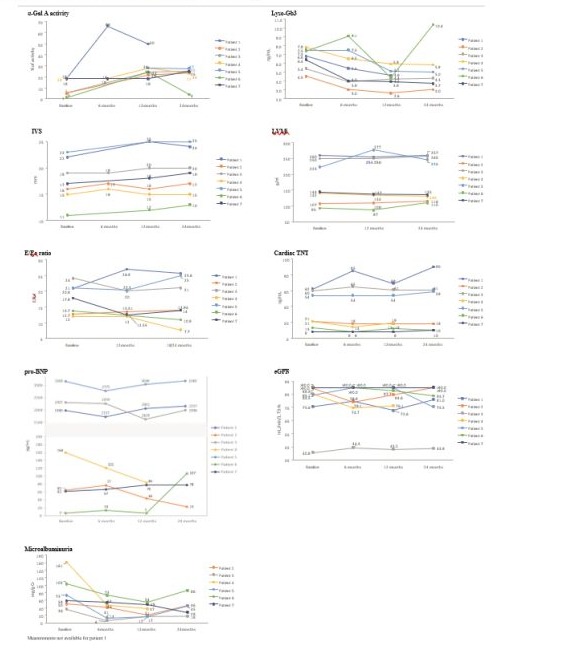
Figure 1: Effect of the switch from ERT to migalastat. Evolution of α-Gal A, Lyso-Gb3, eGFR, microalbuminuria, IVS, LVMi, E/Ea ratio, cardiac TnT, proBNP after 2 years of migalastat. α‐Gal A activity expressed as a percentage of normal. Normal white blood cell activity (µmol/L/h): ≥15.3. α-Gal A, α alpha galactosidase A; Cr, Creatinine; E/Ea, Mitral peak Doppler E-wave to peak mitral annulus velocity ratio; eGFR, estimated glomerular filtration rate; IVS, interventricular septum; LVMi left-ventricular mass index; Lyso-Gb3, deacylated globotriaosylsphingosine; proBNP, pro–brain natriuretic peptide; TnT, troponin-T; UACR, Urine Albumin-to-Creatinine Ratio.
|
Patient |
Age at diagnosis (years) |
Age at switch (years) |
Time since diagnosis (years) |
Typical signs of Fabry Disease |
|
1 |
58 |
62 |
4 |
Hypoacusis |
|
2 |
37 |
54 |
17 |
Hypoacusis |
|
3 |
48 |
64 |
16 |
Neuropathic pain |
|
4 |
42 |
54 |
12 |
- |
|
5 |
52 |
67 |
15 |
Neuropathic pain |
|
6 |
38 |
43 |
5 |
- |
|
7 |
42 |
47 |
5 |
- |
Table 1: Patient demographics and medical history characteristics at switch.
|
Patient characteristics |
Patient 1 |
Patient 2 |
Patient 3 |
Patient 4 |
Patient 5 |
Patient 6 |
Patient 7 |
|||||||
|
T1 |
T2 |
T1 |
T2 |
T1 |
T2 |
T1 |
T2 |
T1 |
T2 |
T1 |
T2 |
T1 |
T2 |
|
|
Time in treatment (months) |
26 |
26 |
120 |
46 |
131 |
35 |
69 |
30 |
137 |
42 |
28 |
26 |
23 |
33 |
|
α-Gal A activity (%) |
<18 |
<18 |
<18 |
<5 |
<18 |
<5 |
<18 |
<18 |
Decreased ‡ |
NA |
<18 |
1 |
<18 |
<18 |
|
Plasma Lyso-Gb3 (ng/mL) |
9.0 |
6.8 |
5.2 |
4.5 |
8.2 |
5.4 |
NA |
7.8 |
NA |
7.5 |
14.5 |
7.4 |
7.5 |
6.4 |
|
eGFR (mL/ min/1.73m2) |
81.5 |
75.6 |
81.4 |
89.5 |
NA |
40.8 |
NA |
85.1 |
116 |
84.5 |
>90 |
90.0 |
>90 |
90.0 |
|
Microalbuminuria (ACR) (mg/g) |
381 |
NA |
15 |
50 |
NA |
36 |
47 |
161 |
NA |
73 |
44 |
103 |
32 |
58 |
|
LVMi (g/m²) |
245 |
260 |
103 |
107 |
194 |
250 |
89 |
142 |
199 |
223 |
102 |
93 |
160 |
144 |
|
IVS (mm) |
23 |
22 |
14 |
16 |
18 |
19 |
16 |
15 |
18 |
23 |
13 |
11 |
17 |
17 |
|
E/Ea |
20.7 |
20.8 |
10 |
12.7 |
12 |
24 |
10.8 |
12 |
23 |
21 |
9.5 |
13.7 |
18 |
17.8 |
|
TnT (ng/L) |
85 |
62 |
85 |
21 |
NA |
60 |
NA |
21 |
NA |
54 |
NA |
13 |
8 |
8 |
|
proBNP (pg/mL)) |
1727 |
1980 |
1727 |
65 |
NA |
2307 |
NA |
160 |
NA |
3160 |
NA |
7 |
63 |
62 |
|
MRI |
LGE |
normal |
NA |
normal |
NA |
normal |
normal |
|||||||
|
Other findings |
kidney transplant (year 2007) |
|||||||||||||
|
†When data was not available at ERT start, the first data available during ERT has been included; ‡Numerical data was missing; the medical record only stated that the enzyme activity levels were decreased. α‐Gal A activity expressed as a percentage of normal. Normal white blood cell activity: ≥15.3 µmol/L/h; Plasma Lyso-Gb3, normal: ≤1.8 ng/mL; eGFR, normal: >90 mL/min/1.73 m2; Microalbuminuria: 30 to 300 mg/g; LVMi, normal: 115 g/m2; IVS normal ≤12 mm; E/Ea < 14; Cardiac TnT, normal: <0.04 ng/L; proBNP, normal: <125 pg/mL. ACR, urine albumin-to-creatinine ratio; α-Gal A, alpha galactosidase A; Cr, Creatinine; E/Ea, Mitral peak Doppler E-wave to peak mitral annulus velocity ratio; eGFR, estimated glomerular filtration rate; IVS, interventricular septum; LGE, late gadolinium enhancement; LVMi left-ventricular mass index; Lyso-Gb3, deacylated globotriaosylsphingosine; MRI, magnetic resonance imaging; NA, not available; proBNP, pro–brain natriuretic peptide; TnT, troponin-T. |
||||||||||||||
Table 2: Clinical and biochemical findings in the seven patients at ERT start/treatment period (T1)† and at migalastat start (T2).
Discussion
Renal function and echocardiographic parameters of seven patients with FD remained stable during the 24-months following the switch from agalsidase alfa ERT to migalastat, suggesting maintenance of disease stability. Overall, the treatment switch was well tolerated. Our results align with Riccio et al.’s study on seven male Fabry patients switching to migalastat for one year, showing no renal or cardiac changes [8]. On the one hand, stable renal function, measured by eGFR, was maintained with no increase in microalbuminuria or proteinuria (data not shown). The kidney-transplanted patient’s renal and cardiac function remained stable after 2 years of migalastat with no need for adjustment of immunosuppressive treatments, suggesting safety, though it has not been specifically tested in kidney transplant recipients. The case aligns with a prior report of a kidney transplant patient achieving stability with migalastat therapy for one year [11]. However, further research is warranted to explore migalastat’s efficacy and safety in these patients. On the other hand, no LVH progression was observed. Unlike prior studies [7,12], only two patients showed a reduction in ventricular mass with this therapy. Our patients had cardiac damage on migalastat start, making it difficult to detect a decrease in ventricular mass. Troponin T and NT-proBNP showed no significant changes following migalastat treatment. Nevertheless, myocardial fibrosis irreversibility emphasizes the need for early treatment initiation [13]. Long-term ERT may slow disease progression, yet issues often arise [14]. Despite a high initial disease burden, two years of migalastat maintained stable cardiac and renal function, suggesting a potential benefit.
Our findings are also consistent with the conclusion of Müntze et al. that higher enzyme activity tends to lower Lyso-Gb3 levels. [7]. However, to date, no biomarker has been validated to measure treatment response to migalastat. Therefore, all biochemical and clinical parameters should be considered when monitoring patients [15].
These cases could help clinicians worldwide in treatment decisions for this mutation and emphasize the importance of a multidisciplinary approach. Since FD is a rare disease whose clinical manifestations and severity are highly influenced by the underlying GLA mutation [16], the description of seven clinical cases with the same mutation is noteworthy. As in other studies, the number of patients with the same mutation is smaller, supporting the data presented here [7,17].
A growing number of doctors are using migalastat for FD, both in treatment-naïve patients and in patients switching from ERT. According to a recent review of publications, only five studies have examined the safety and efficacy of switching from ERT to migalastat to date [18]. This case series provides valuable insights, especially considering the advanced stage of the described patients, including one transplant recipient.
In conclusion, migalastat preserved renal and cardiac parameters and increased enzyme activity in our Fabry patients with the S238N mutation. This treatment switch was safe and well tolerated, in line with the findings from pivotal and long-term studies. Coordinated multidisciplinary follow-up is crucial in the real-world setting, highlighting the need for further research to enhance the clinical management of FD.
Author contribution statement: Conceptualization: RN, RSM, VCP, and EGP; methodology: RN, RSM, VCP, and EGP; validation: RN, RSM, VCP, and EGP; investigation: RN, RSM, VCP, and EGP; resources: RN, RSM, VCP, and EGP; data curation: RN, RSM, VCP, and EGP; writing: RN, RSM and VCP; review and editing: RN, RSM, VCP, and EGP; visualization: RN, RSM and VCP; and supervision: RN, RSM and VCP. All authors approved the final version of the manuscript.
Acknowledgements: Medical editing support was provided by María Yuste at Evidenze Health España, S.L.U. during the preparation of this paper, funded by Amicus Therapeutics. Responsibility for opinions, conclusions and interpretation of data lies with the authors.
Conflict of Interest: Raúl Noguera received honoraria for participating on advisory boards and received support for attending meetings and/or for travel from Shire, Takeda, Amicus Therapeutics, and Sanofi-Genzyme. Rosario Sánchez Martínez received honoraria for participating on advisory boards and in clinical trials and received support for attending meetings and/or for travel from Shire, Takeda, Amicus Therapeutics, Sanofi-Genzyme, Chiesi, EusaPharma, and Bayer. Vicente Climent-Payá received honoraria for participating on advisory boards and in clinical trials and received support for attending meetings and/or for travel from Shire, Takeda, Amicus Therapeutics, and Sanofi-Genzyme. Elena García-Payá reports no conflict of interest. Amicus Therapeutics funded the medical editing of the manuscript.
Data availability: Data available on request from the authors.
References
- Ortiz, A, Germain, D. P, Desnick, R. J, Politei, J, Mauer, M, et al (2018) Fabry Disease Revisited: Management and Treatment Recommendations for Adult Patients. Mol Genet Metab, 123: 416-427.
- Colon, C, Ortolano, S, Melcon-Crespo, C, Alvarez, J. V, Lopez-Suarez, O. E, et al (2017) Newborn Screening for Fabry Disease in The NorthWest of Spain. Eur J Pediatr, 176: 1075-1081.
- Spada, M, Pagliardini, S, Yasuda, M, Tukel, T, Thiagarajan, G, et al (2006) High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening. Am J Hum Genet, 79: 31-40.
- Monserrat, L, Gimeno-Blanes, J. R, Marín, F, Hermida-Prieto, M, García-Honrubia, A, et al (2007) Prevalence of Fabry Disease in A Cohort Of 508 Unrelated Patients with Hypertrophic Cardiomyopathy. J Am Coll Cardiol, 50: 2399-403.
- Barba-Romero, M, Serena, J, Puig, J. M, Valverde, C. V, Climent, V, et al 2019. Clinical Profile of Women Diagnosed with Fabry Disease Non-Receiving Enzyme Replacement Therapy. Med Clin (Barc), 153: 47-55.
- Payá Mora, E. (2017) Characteristics of Anderson-Fabry Disease Caused by The S238n Mutation of The Gla Gene In the Health Department of Elda. Universidad Miguel Hernández.
- Müntze, J, Gensler, D, Maniuc, O, Liu, D, Cairns, T, Oder, D, Hu, K, Lorenz, K, Frantz, S, Wanner, C. & Nordbeck, P. et al (2019) Oral Chaperone Therapy Migalastat For Treating Fabry Disease: Enzymatic Response and Serum Biomarker Changes After 1 Year. Clin Pharmacol Ther, 105, 1224-1233.
- Riccio, E, Zanfardino, M, Ferreri, L, Santoro, C, Cocozza, S, et al (2020) Switch from Enzyme Replacement Therapy to Oral Chaperone Migalastat For Treating Fabry Disease: Real-Life Data. European Journal of Human Genetics, 28: 1662-1668.
- Levey, A. S, Coresh, J, Greene, T, Stevens, L. A, Zhang, Y, et al (2006) Using Standardized Serum Creatinine Values in The Modification of Diet in Renal Disease Study Equation for Estimating Glomerular Filtration Rate. Annals of Internal Medicine, 145: 247-254.
- Levey, A. S, Stevens, L. A, Schmid, C. H, Zhang, Y. L, Castro, A. F, et al (2009) A New Equation to Estimate Glomerular Filtration Rate. Ann Intern Med, 150, 604-12.
- Di Stefano, V, Mancarella, M, Camporeale, A, Regalia, A, Ferraresi, M, et al (2021) Migalastat Treatment in A Kidney-Transplanted Patient with Fabry Disease and N215s Mutation: The First Case Report. Pharmaceuticals (Basel), 14.
- Hughes, D. A, Nicholls, K, Shankar, S. P, Sunder-Plassmann, G, Koeller, D, et al (2017) Oral Pharmacological Chaperone Migalastat Compared with Enzyme Replacement Therapy in Fabry Disease: 18-Month Results from The Randomised Phase Iii Attract Study. J Med Genet, 54: 288-296.
- Weidemann, F, Niemann, M, Breunig, F, Herrmann, S, Beer, M, et al (2009) Long-Term Effects of Enzyme Replacement Therapy on Fabry Cardiomyopathy. Circulation, 119, 524-529.
- Arends, M, Biegstraaten, M, Hughes, D. A, Mehta, A, et al (2017) Retrospective Study of Long-Term Outcomes Of Enzyme Replacement Therapy In Fabry Disease: Analysis Of Prognostic Factors. Plos One, 12, E0182379.
- Bichet, D. G, Hopkin, R. J, Aguiar, P, Allam, S. R, Chien, Y. H, et al (2023) Consensus Recommendations for The Treatment and Management Of Patients With Fabry Disease On Migalastat: A Modified Delphi Study. Front Med (Lausanne), 10, 1220637.
- Germain, D. P. 2010. Fabry Disease. Orphanet J Rare Dis, 5, 30.
- Lenders, M, Nordbeck, P, Kurschat, C, Eveslage, M, Karabul, N, et al (2022) Treatment of Fabry Disease Management with MigalastatOutcome from A Prospective 24 Months Observational Multicenter Study (Famous). Eur Heart J Cardiovasc Pharmacother, 8, 272-281.
- Perretta, F. & Jaurretche, S. (2023) Fabry Disease: Switch from Enzyme Replacement Therapy to Oral Chaperone Migalastat: What Do We Know Today? Healthcare (Basel), 11.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.