Medullary Thyroid Carcinoma Literature Review and Current Management
by Rodrigo Arrangoiz*, Fernando Cordera, David Caba, Eduardo Moreno, Enrique Luque de-Leon, Manuel Muñoz
Department of Surgical Oncology, Sociedad Quirúrgica at the American British Cowdray Medical Center, Mexico
*Corresponding author: Rodrigo Arrangoiz, Department of Surgical Oncology, Sociedad Quirúrgica at the American British Cowdray Medical Center, Mexico. Tel: +525516647200; Email: rodrigo.arrangoiz@gmail.com
Received Date: 29 August, 2018
Accepted Date: 21 September, 2018
vzPublished Date: 27 September, 2018
Citation: Arrangoiz R, Cordera F, Caba D, Moreno E, Luque de-Leon E, et al. (2018) Medullary Thyroid Carcinoma Literature Review and Current Management. J Clin Endocrinal and Diabetes: JCED-118. DOI: 10.29011/ JCED-118/100018
1. Abstract
Medullary Thyroid Cancer (MTC) accounts for approximately 1.7% of all thyroid malignancies. The vast majority of MTC are sporadic, but 15% to 25% of the cases result from a germline mutation in the RET proto-oncogene. Familial Medullary Thyroid Cancer (FMTC) can be seen as an element of the Multiple Endocrine Neoplasia Syndrome (MENS) type 2A or 2B or as part of FMTC. This review discusses the contemporary approaches available for the evaluation and diagnosis of a patient with believed to have MTC. The current therapeutic approach to MTC is chiefly surgical, consisting of a total thyroidectomy and lymph node dissection. The degree and timing of the surgery are discussed centered on evidence-based medicine. Systemic treatment options are limited for MTC, but some therapeutic targets show promise for the development of new treatment option in the near future.
In this manuscript, we discuss the epidemiology, pathophysiologic mechanisms, diagnostic challenges and the innovative therapeutic interventions of diabetic neurotrophic keratopathy, an entity with potentially serious implications for diabetic patients, both type 1 as well as type 2 alike.
2. Introduction
In 1959 Hazard, Hawk, and Crile provided the first histologic description of Medullary Thyroid Carcinoma (MTC), but they were not the first to identify MTC, due to the fact that more than a century before, Jacquet had described a thyroid tumor with amyloid [1,2]. Williams identified that MTC originated from the neural crest derived perifollicular cell or C-cells of the thyroid gland [3] and Tashjian et al. discovered that the C-cells secreted calcitonin [4]. Soon after the identification that MTC represented a unique thyroid malignancy, it was recognized that these tumors occurred either sporadically or in a hereditary form as a component of the type 2 Multiple Endocrine Neoplasia (MEN) syndromes, MEN2A (Sipple syndrome) and MEN2B (Wagenmann-Froboese syndrome), and the related syndrome, familial MTC (FMTC). Comprehensive surgical resection of the primary and nodal metastases by compartment-oriented resection has been the goal standard of treatment. Resection of residual and recurrent disease successfully reduces calcitonin levels and controls complications of disease in the central neck. Radioactive iodine and conventional chemotherapy have not been shown to be very effective. External beam radiation therapy has a role, but it is limited, in the armamentarium against this disease. Novel systemic treatments, with small molecule inhibitors, hold promise and are being tested in clinical trials for patients with metastatic disease.
2.1. Epidemiology and Etiology
Medullary thyroid carcinoma is a neuroendocrine tumor of the para-follicular cells or C cells of the thyroid gland [5]. Approximately 1.7% of all thyroid neoplasms are medullary carcinomas [5,6]. Although most cases are sporadic, 15% to 25% of the cases are part of an autosomal dominant hereditary syndrome [5].
The production of calcitonin is a characteristic feature of this tumor [7,8]. The C cells originate in the embryonic neural crest; as a result, medullary carcinomas often have the clinical and histological characteristics of other neuroendocrine tumors such as carcinoids and pancreatic islet cell tumors.
The RET proto-oncogene (RE- arranged during Transfection) was discovered in 1985 by Takahashi et al. [9]. The RET proto-oncogene is expressed in cells derived from the neural crest, the branchial arches, and the urogenital system [10,11]. It is located on chromosome 10q11.2, encodes a single-pass transmembrane receptor of the tyrosine kinase family. Soon after this discovery it was found that nearly all patients with MEN2A, MEN2B, and FMTC have RET germline mutations and roughly 50% of sporadic MTCs have somatic RET mutations [12-16]. Researchers recently revealed that 18% to 80% of sporadic MTCs lacking somatic RET mutations have somatic mutations of HRAS, KRAS, or rarely NRAS [17-19].
The probability that an apparently sporadic case of MTC is familial should be considered preoperatively. Despite the absence of a family history of MTC, germline mutations in the RET proto-oncogene have been identified in less than 10% of cases of a seemingly sporadic MTC (index cases) [20,21].
In sporadic MTC the somatic RET codon M918T mutation appears to indicate an aggressive clinical course and a poor prognosis [22,23]. Romei C et al. reviewed 160 patients with sporadic MTC and found that the prevalence of somatic RET codon M918T mutations varied depending on tumor size: < 1 cm, 11.3% of the cases; 1 to 2 cm, 11.8% of the cases; 2 to 3 cm, 31.8% of the cases; and > 3 cm, 58.8% of the cases [24].
The results of these study raise the question of whether RET acts singlehandedly as the initiator of oncogenesis in sporadic MTC or is activated later as a driver of tumor growth, with other genes playing a significant role in MTC onset. Another reason for these findings is that M918T mutated tumors have a higher growth rate and are more likely to be identified when they are bigger. The low prevalence of the M918T mutation in micro carcinomas may signify a different entity such as carcinoma in situ; precisely because it is not driven by RET.
Several guidelines for the management of sporadic and hereditary MTC have been published by several groups including the North American Neuroendocrine Tumor Society, the National Comprehensive Cancer Network, and the American Thyroid Association (ATA) [5,25-27]. These guidelines described the disease phenotypes associated with specific RET mutations in hereditary MTC and recommended timing of early thyroidectomy based on the specific RET mutation [5,25-27]. Three of the guidelines used either the TNM designation of the American Joint Committee on Cancer (AJCC), or terms such as Level I, II, or III, or ‘‘high,’’ ‘‘higher,’’ or ‘‘highest,’’ to allocate progressive increases in aggressiveness of the MTC [25,26,28].
The criteria used to categorize the aggressiveness of the disease was the development of MTC at an early age, frequently in association with metastatic disease. The original ATA guidelines used A, B, C, and D designations to define categories of RET mutations associated with increasing aggressiveness of the MTC [25].
Due to significant misunderstanding regarding the different ATA risk categories, the task force recommends that the A and B categories be combined into a new category, ‘‘moderate risk’’ (MOD); category C be changed to a new category, ‘‘high risk’’ (H); and category D be changed to a new category, ‘‘highest risk’’ (HST) [recommendation # 1 from the ATA guidelines) [5].
The ATA-HST category includes patients with MEN2B and the RET codon M918T mutation, the ATA-H category includes patients with RET codon C634 mutations and the RET codon A883F mutation, and the ATA- MOD category includes patients with RET codon mutations other than M918T, C634, and A883F [5]. Ever since the discovery of the RET oncogene, over 100 mutations, duplications, insertions, or deletions involving RET proto-oncogene have been identified in patients with hereditary MTC.
2.2. Hereditary Medullary Thyroid Carcinoma
More than 100 years have passed since MTC was noted as a separate disease entity within the broad spectrum of malignant thyroid tumors [1,29,30]. By the first half of the 20th century, the unusual tumor biology of the sporadic type had been described as “small tumors with early lymphatic and hematogenous spread, whereas metacentric MTC was being recognized as the hallmark of hereditary tumors [31].
Possibly the first report of MEN 2B was written by Walther Burk in 1901 [29]. Nonetheless, in 1965 E.D. Williams defined the syndromic MTC as a separate entity by publishing the first comprehensive series of hereditary pheochromocytoma associated with MTC [32]. In 1968 Steiner and colleagues described a family with the concurrence of MTC, pheochromocytoma, hyperparathyroidism (HPTH), and Cushing’s syndrome [33].
They suggested that the entity be named MEN2 in contradistinction to the earlier described hereditary disease, MEN1 [33,34]. The syndrome that Steiner et al. described is now known as MEN2A (incidence 1/1,973,500) [35]. In retrospect the earliest documented family with MEN2A was from Sweden and concerned a kindred dating to the early 1700s [36].
The disease spectrum of MEN2A has expanded to include two variants: patients with associated cutaneous lichen amyloidosis (CLA) and patients with associated Hirsch sprung disease (HD) [37,38]. The MEN2B syndrome (incidence 1/ 38,750,000) variably described by Williams and Pollock, Schimke and colleagues, and Gorlin and associates, accounts for 5% of hereditary MTCs [35,39-41].
Patients with MEN2B develop MTC and pheochromocytoma and exhibit a recognizable phenotype. Farndon et al. described FMTC [42,48]. Initially, very strict criteria defined the diagnosis of FMTC: more than 10 family members with MTC, multiple carriers or affected members over 50 years of age, and an adequate medical history (particularly in older family members) to exclude the presence of pheochromocytoma, and HPTH [42]. A less strict definition was the presence in at least four family members of MTC without other manifestations of MEN2A [43].
At this time, the opinion of most investigators is that FMTC should not be a freestanding syndrome; rather it should represent a variant along the spectrum of disease expression in MEN2A [5]. The Task Force agrees that FMTC should not be defined as a form of hereditary MTC distinct from MEN2A and MEN2B [28,43]. Rather it should be recognized as a variant of MEN2A to include families with only MTC who meet the original strict criteria for FMTC, small families of at least two generations with at least two, but less than 10, subjects with RET germline mutations, small families in which two or fewer members in a single generation have RET germline mutations, and single individuals with a RET germline mutation [5]. Based on available evidence there should be two MEN2 syndromes: MEN2A and MEN2B. Within MEN2A, which accounts for 95% of MEN2 cases, there should be four variants: classical MEN2A (represented by the uniform presence of MTC and the less frequent occurrence of pheochromocytoma, or HPTH, or both), MEN2A with CLA, MEN2A with HD, and FMTC (recommendation # 2 of the ATA guidelines [5]. (Table 1) summarizes the clinical features of MEN type 2.
2.3. Clinical Presentation
The sporadic form of medullary thyroid cancer typically presents as a unilateral solitary nodule (75% to 95% of patients) in the fifth decade of life [31,44-46]. The family forms, such as MEN 2A, MEN 2B and familial medullary cancer, occur in the fourth decade of life and are typically multifocal [31,44-46].
Due to the embryological origin of MTC (the cells C), these tumors are located in the upper poles of the thyroid gland where these cells reside [7,8]. Approximately 50% to 70% of patients with MTC have clinically detectable cervical lymph node involvement at the time of diagnosis [5,7,8], roughly 15% have symptoms of compression or invasion of the upper aero digestive tract, such as dysphagia or hoarseness, and approximately 5% to 10% have distant metastasis [7,47].
Hormonal secretion by the tumor may produce systemic symptoms. Tumor secretion of calcitonin, calcitonin gene-related peptide, or other substances can cause diarrhea or facial flushing in patients with advanced disease. In addition, occasional tumors may secrete corticotrophin (ACTH), causing ectopic Cushing's syndrome [44]. Basal serum calcitonin concentrations usually show a direct relationship with tumor volume but also echo tumor differentiation, and they are almost always elevated in patients with a palpable tumor [48].
The majority of MTCs also secrete carcinoembryonic antigen (CEA), which, like calcitonin, can be used as a tumor marker [49,50]. In addition, the expression of CEA on MTC cells has led to the use of anti-CEA antibodies for immunotherapy. Patients with MTC have normal thyroid function test.
There are several ultra-sonographic features of thyroid nodules (eg, solid hypoechoic, irregular margins [infiltrative, micro-lobulated], micro calcifications), taller than wide shape, rim calcifications with small extrusive soft tissue component, evidence of extra thyroidal extension (ETE) that are associated with a higher cancer [51]. Nevertheless, there are no ultra-sonographic features that are pathognomonic for thyroid cancer. Moreover, the preponderance of studies evaluating suspicious ultrasound features of nodules focused on papillary thyroid cancer (PTC). In a retrospective series evaluating the ultrasound features of nodules that were histologically proven to be MTC and PTC, 50% of MTCs were solid and hypoechoic and 16% showed micro calcifications, compared with 69.2% and 69.2%, respectively, for PTC [52]. In other study, hypoechoic nodules were identified in 50% to 89% of the cases and micro calcifications in 30% to 70% of the cases of MTC [53,54]. Macro calcifications are identified in 16% to 30% of the cases of MTC [52,54,55]. The diagnosis of MTC is rarely suggested by the presence of dense calcifications seen on radiographs or imaging of the anterior neck.
Approximately 50% to 70% of patients with medullary thyroid cancer have clinically detectable cervical lymph node involvement at the time of diagnosis [5,7,8], roughly 15% have symptoms of compression or invasion of the upper aero digestive tract, such as dysphagia or hoarseness, and approximately 5% to 10% have distant metastasis [7,47]. Survival of patients with MTC lies in between that of differentiated thyroid cancers and undifferentiated (anaplastic) thyroid cancers. When the disease is limited to the thyroid gland, the 10-year survival rate is 90% versus patients with distant metastatic disease that has a 10-year survival of only 20% [8].
Survival of patients with MTC lies in between that of differentiated thyroid cancers and undifferentiated (anaplastic) thyroid cancers. When the disease is limited to the thyroid gland, the 10-year survival rate is 90% versus patients with distant metastatic disease that has a 10-year survival of only 20% [8].
2.4. Pathology
Sporadic MTC typically presents as a single circumscribed but non-encapsulated, gray-tan tumor (Figure 1). In sporadic tumors, approximately 75% to 95% are solitary, and 5% to 25% are bilateral or multifocal [31,44-46]. In familial forms, 94% are bilateral or multifocal, and 6% are solitary [31,44-46].
Macroscopically MTC tumors are usually solid, gray-tan-yellow, firm, may be infiltrative. Larger lesions may have hemorrhage and necrosis. Tumors that measure in their greatest dimension less than 1 cm in size are called micro carcinomas [56]. Histologically, the cells are uniform polygonal cells with fine granular eosinophilic cytoplasm with central nuclei (Figure 2). Variable numbers of spindle cells are present in nearly all tumors. The presence of amyloid is considered to be a distinctive feature of MTC, although it may not necessarily be found in all cases (Figure 3) [57]. The amyloid differs from that of other tumors in that it is formed from calcitonin or procalcitonin molecules.
Histologically, MTC can be classified based on the dominant histologic pattern. These categories a broad but the most common include the classic, amyloid-rich, trabecular, and amphicrine variants (Table 1). Classic variants are most common (48.9%), followed by the amyloid-rich variants (38.3%) [44,58]. C-cell hyperplasia is associated with MTC, particularly in the familial forms. It is believed that the presence of C cell hyperplasia is an omen for the development of hereditary medullary cancer [7,31,44-46,59,60].
These tumors are not encapsulated, nor well-defined, and consist of a heterogeneous mixture of spindle or round cells [7,31,44-46]. The cells are separated by fibrous septa and amyloid, the last of which helps in the diagnosis by means of an immunohistochemical stain for calcitonin and carcinoembryonic antigen [7]. Although these tumors grow slowly, they have a tendency, to metastasize early, usually before the primary tumor has reached 2 cm in diameter [7,8].
The evaluation of a thyroid neoplasm with any features reminiscent of an MTC should include Immunohistochemistry (IHC) analysis to determine the presence of markers such as calcitonin, chromogranin, and CEA and the absence of thyroglobulin (recommendation # 16 ATA guidelines) [5].
Comprehensive documentation of the pathologic features of every MTC should follow the synoptic reporting guidelines of the College of American Pathologists Protocol for the Examination of Specimens from Patients with Carcinomas of the Thyroid Gland (recommendation # 17 ATA guidelines) [5]. Patients with MTC, a complete morphological examination of the total thyroid gland is recommended to determine the presence of C cell hyperplasia or multifocal neoplasia (recommendation # 18 ATA guidelines) [5].
2.5. Diagnosis
Fine-Needle Aspiration (FNA) biopsy is used to make the diagnosis of MTC in patients with a solitary thyroid nodule (or a dominant nodule within a multinodular goiter). The sensitivity of FNA for the diagnosis of MTC is 50% to 80%, though a higher sensitivity can be achieved by adding the immunohistochemical staining for calcitonin [61,62]. When the suspicion for MTC is high (patient with flushing, diarrhea, in the context of a thyroid nodule), calcitonin can be measured in the washout of the FNA biopsy needle [63].
The diagnosis of MTC can be made postoperatively after a thyroid lobectomy for a suspicious or indeterminate FNA biopsy. Surgical specimens from patients with MTC show spindle-shaped and frequently pleomorphic cells without follicle development because these cells originate from the calcitonin-producing perifollicular C cells of the thyroid [57].
The routine uses of serum calcitonin screening in patients with thyroid nodules is controversial and the ATA guidelines cannot recommend in favor or against it use (recommendation # 4 ATA guidelines) [51].
The elevated incidence of falsely elevated serum calcitonin levels and the inability to confirm the elevated calcitonin by the pent gastrin stimulation in the United States argue against its use. Additionally, some patients with loco regional metastases or locally invasive MTC will have normal unstimulated serum calcitonin concentrations [51]. Elevated calcitonin levels may also be seen in patients with hypergastrinemia, hypercalcemia, renal insufficiency, neuroendocrine neoplasms, papillary and follicular thyroid carcinomas, goiter, and in chronic autoimmune thyroiditis [64,65]. Likewise, protracted use of omeprazole (greater than two to four months), beta blockers, and glucocorticoids has been linked with hypercalcitoninemia [66].
The presence of heterophilic antibodies to calcitonin can falsely raise calcitonin levels [67]. Carcinoembryonic antigen (CEA) levels can be found to be elevated in patients with heterophilic antibodies, gastrointestinal tract inflammatory disease, benign lung disease, non-thyroid malignancies, and cigarette smoking [5].
Physicians should be cognizant of falsely elevated or low serum calcitonin levels that might occur in a variety of clinical diseases other than MTC. They should consider this possibility when serum calcitonin levels are disproportionate to the expected clinical findings (recommendation # 13 ATA guidelines) [5]. Physicians should be aware that calcitonin levels are significantly elevated in children under three years of age (especially under six months of age). Similarly, calcitonin concentrations are higher in males compared with females (recommendation # 14 ATA guidelines) [5].
Patients with newly diagnosed MTC should be considered to have hereditary MTC until proven otherwise. Hence, patients with MTC should have a full family history taken at the time of initial consultation. Physical examination should take note of the size of the palpable neck nodules, fixation to surrounding structures, and the presence of cervical lymphadenopathy [8]. Distinctive features of the MEN 2B phenotype should also be noted. Clinical manifestations of patients with extensive localized disease include dysphagia, hoarseness, dyspnea, and coughing [8]. Direct examination of the vocal cords prior to surgical intervention is warranted if any symptoms or signs of possible involvement of the recurrent laryngeal nerve are noted during the clinic visit [51]. Patients presenting with elevated levels of calcitonin may exhibit diarrhea as the initial symptom of their disease [5]. Lichen planus amyloidosis may be an early indicator of MEN 2A and should prompt genetic testing and appropriate clinical workup.
Patients diagnosed with MTC via cytological evaluation the initial work-up should include a complete history and physical examination, measurement of serum calcitonin, CEA, ultrasound of the neck, in selected cases calcitonin in FNA washout, genetic testing for germline RET mutations, and biochemical evaluation for coexisting tumors, especially pheochromocytoma (recommendation # 15, 19, 21, 22 ATA guidelines) [5].
Preoperative calcitonin levels may correlate with tumor size in both sporadic and familial cases of MTC [68]. Preoperative calcitonin level of cutoff 50 pg/mL may help predict who will have a biochemical complete response after surgery. Cohen et al. reported in their study in which 45 patients who had a preoperative calcitonin level of 50 pg/mL or less, 44 had normal levels after surgery. Compared to only 50 of 120 patients with preoperative calcitonin levels higher than 50 pg/mL had normal concentrations after surgery. In a second study of 224 patients with MTC, by Machens et al. 62% of the patients without nodal metastases had normal calcitonin postoperatively, while 10% of patients of nodal metastasis had normal postoperative calcitonin levels [69].
Evaluation of calcitonin and CEA doubling times postoperatively provides sensitive markers for progression and aggressiveness of metastatic MTC [70,71]. In a study of 65 patients followed for 3 to 30 years’ postoperative calcitonin doubling time was a prognostic factor for survival [70]. Ten-year survival was 8% for doubling times under six months, 37% for doubling times between six months and two years, and 100% for doubling times greater than two years [70].
Contrast-enhanced CT of the neck and chest, three-phase contrast-enhanced multi-detector liver CT, or contrast-enhanced MRI of the liver, and axial MRI and bone scintigraphy are recommended in patients with extensive neck disease and signs or symptoms of regional or distant metastases, and in all patients with a serum calcitonin greater than 500 pg/mL (recommendation # 22 ATA guidelines) [5]. FDG-PET/CT nor F-DOPA-PET/CT are recommended to detect the presence of distant metastases (recommendation # 23 ATA guidelines) [5]. In advanced MTC, a marked increase in the CEA level out of proportion to a lower calcitonin level or normal or low levels of both calcitonin and CEA indicate poorly differentiated MTC [5]. Our approach is outlined in Figure 4 and is consistent with NCCN and ATA Guidelines for Management of MTC [5,47].
All patients with newly diagnosed C cell hyperplasia or apparently sporadic MTC should have germline RET mutation testing (recommendation # 21 ATA guidelines) [5,47]. Preliminary germline testing in patients with C cell hyperplasia or apparently sporadic MTC should include sequencing of exons 10, 11, and 13 through 16 of the RET gene [5,47,72]. Sequencing of the residual exons in the RET gene should be considered in patients with clinical manifestations or family history indicative of hereditary medullary syndromes who express no mutations in exons 10, 11, or 13 through 16 [72].
We keenly encourage discussion with genetic counselors who are aware of both the ethical issues and legal informed consent requirements that are involved in germline testing [5]. If the index patient is positive for a germline mutation, family members should be offered genetic counseling and genetic screening. Studies of patients with MTC have found, on average, that approximately 6% to 7% (range 1.5% to 24%) have germline RET mutations [21,73-75]. In one study, 7.3% of patients with apparently sporadic MTC had mutations, and in 51% of the cases, it was identified that relatives of the patients with MTC were gene carriers [75]. No previous family history of the familial medullary syndromes was identified in 75% of the cases.
Roughly 60% of patients with sporadic MTC have somatic (acquired) mutations in the RET gene within the tumor cells (Table 2) [76-78]. These mutations are present only in the tumor cells and are not detected by standard genetic testing (leukocyte DNA). The occurrence of somatic RET mutations are associated with lymph node metastases, persistent disease, and lower survival [23]. Nevertheless, in one study, only mutations in exons 15 and 16 of the RET gene correlated with a worse prognosis, while those in other exons had a more indolent course [17].
The vast majority of patients with MTC require biochemical evaluation for coexisting tumors (chiefly pheochromocytoma and hyperparathyroidism) before surgery. For patients with unknown RET mutational status and for patients who have a germline RET mutation, serum calcium levels (to rule out hyperparathyroidism requiring simultaneous surgical intervention) and serum fractionated metanephrines (initial screen for pheochromocytoma) [7,8].
Normal serum fractionated metanephrines dismiss a symptomatic catecholamine-secreting neoplasm, but minimal elevations of normetanephrines could be falsely positive, in which case additional testing including 24-hour urinary fractionated metanephrines, catecholamine’s, and adrenal imaging may be useful to successfully rule in or rule out pheochromocytoma prior to the operation [7]. Adrenal gland imaging should not be performed unless there is biochemical evidence of a potential pheochromocytoma. Patients with a negative RET proto-oncogene testing and no family history of MEN2 syndrome, biochemical testing for coexisting tumors is typically not required.
2.6. Staging
The American Joint Committee on Cancer defines four stages of disease in MTC centered on pathologic clinic pathologic tumor characteristics (eighth edition) based upon tumor size and the presence or absence of extra thyroidal invasion, local and regional nodal metastases, and distant metastases (Table 3,4) [79]. An analysis of the National Cancer Database and the Surveillance, Epidemiology, and End Results (SEER) data from patients with MTC demonstrated that the seventh and eighth editions of the AJCC staging system were associated with five-year overall survival rates of 95% for stage I, 91% in stage II, 89% in stage III, and 68% in stage IV. Additionally, disease-specific survival rates were 100% in stage I, 99% in stage II, 97% in stage III, and 82% in stage IV [80].
2.7. Treatment
MTC can be cured only by complete resection of the thyroid tumor and any local and regional metastases [5,47]. For patients with residual or recurrent disease after primary surgery or for those with distant metastases, the most suitable treatment (surgery, chemotherapy, or radiotherapy) is less clear.
For MTC limited to the neck and no evidence of involved cervical lymph nodes on preoperative ultrasound, total thyroidectomy with prophylactic bilateral central compartment lymph node dissection is the desired initial treatment (recommendation # 24 ATA guidelines) [5,47]. Around 10% of patients with sporadic MTC and all patients with familial MTC have bilateral or multifocal disease [5]; furthermore, the latter all have premalignant diffuse C cell hyperplasia. Hence, total thyroidectomy rather than unilateral lobectomy is the preferred surgical approach [5]. Some authors, like Machens et al. suggest that prophylactic central compartment neck dissection is not required in patients with small intrathyroidal MTCs with a preoperative calcitonin less than 20 pg/mL, as metastatic lymph nodes are exceedingly rare in these circumstances [81].
Controversy exists if patients with MTC and no evidence of lymph node metastases in the neck on ultrasound, and no distant metastases, if lymph node dissection of the lateral compartments (levels II to V) should be considered based on serum calcitonin levels (recommendation # 25 ATA guidelines) [5].
The ATA guidelines could not attain a consensus agreement on this topic but did recommend that prophylactic lateral neck dissections "may be considered based on serum calcitonin levels" (recommendation # 25 ATA guidelines) [5].
Some of the panelist did not recommended routine prophylactic lateral neck dissections if there was no evidence of disease on preoperative neck ultrasound. Nonetheless, some members suggested in utilizing preoperative calcitonin values to guide the extent of prophylactic neck dissections by recommending prophylactic ipsilateral central and ipsilateral lateral neck dissection for patients with basal serum calcitonin values greater than 20 pg/mL and prophylactic dissection of uninvolved contralateral lateral neck compartments for serum calcitonin greater than 200 pg/mL [5].
Patients with MTC limited to the neck and cervical lymph nodes should have a total thyroidectomy, dissection of the central compartment lymph nodes (level VI), and dissection of the involved lateral neck compartments (levels II to V). When preoperative imaging is positive in the ipsilateral lateral neck compartment but negative in the contralateral neck compartment, contralateral neck dissection should be considered if the basal serum calcitonin level is greater than 200 pg/mL (recommendation # 26 ATA guidelines) [5].
In the presence of extensive regional or metastatic disease less aggressive surgery in the central and lateral neck may be appropriate since the goals of surgery is largely palliative in order to preserve speech, swallowing, parathyroid function, and shoulder mobility. External Beam Radiotherapy (EBRT), systemic medical therapy, and other nonsurgical therapies should be considered to achieve local tumor control (recommendation # 27 ATA guidelines) [5]. In the presence of grossly invasive disease, more aggressive procedures with resection of involved neck structures may be appropriate in properly selected patients, but function-preserving (speech, swallowing) approaches are preferred. Disfiguring radical neck dissections do not improve prognosis and are not indicated. The surgical approach should be individualized based upon the patient's wishes, life expectancy, and other medical comorbidities
Following unilateral lobectomy for presumed sporadic MTC a completion thyroidectomy is recommended in patients with a RET germline mutation, an elevated postoperative serum calcitonin level, or imaging studies indicating residual MTC. The presence of an enlarged lymph node in association with a normal serum calcitonin level is not an indication for repeat surgery (recommendation # 28 ATA guidelines) [5]. Patients who underwent a poor lymph node dissection at the initial thyroidectomy a repeat operation, including compartment oriented lymph node dissection, should be considered if the preoperative basal serum calcitonin level is less than 1000 pg/mL and five or less metastatic lymph nodes were removed at the initial surgery (recommendation # 29 ATA guidelines) [5].
Patients with MTC treated with a total thyroidectomy, parathyroid glands should be preserved in situ on a vascular pedicle. If all normal parathyroid glands are resected or if none appear viable at the end of the procedure, slivers of a parathyroid gland should be transplanted into the sternocleidomastoid muscle in patients with sporadic MTC, MEN2B, or MEN2A and a RET mutation rarely associated with Primary Hyperparathyroidism (PHPT). In patients with MEN2A and a RET mutation associated with a high incidence of PHPT, the parathyroid tissue should be transplanted in a heterotopic muscle bed, such as the brachioradialis muscle in the non-dominant arm (recommendation # 30 ATA guidelines) [5].
Skilled physicians and surgeons in tertiary care centers should be responsible for the management of children with MEN2A or MEN2B (recommendation # 33 ATA guidelines) [5]. Children in the ATA-HST category with a RET codon M918T mutation should have a thyroidectomy in the first year of life, conceivably even in the first months of life (recommendation # 34 ATA guidelines) [5]. In the absence of suspicious lymph nodes, the performance of a central neck dissection should be based on whether the parathyroid glands can be recognized and left in situ or auto transplanted. The surgeon and pediatrician caring for the patient, in consultation with the child’s parents, should decide the timing of thyroidectomy (recommendation # 34 ATA guidelines) [5].
Children in the ATA-H category should undergo a thyroidectomy at age 5 years, or earlier based on the detection of elevated serum calcitonin concentrations (recommendation # 35 ATA guidelines) [5]. A central neck dissection should be performed in children with serum calcitonin levels above 40 pg/mL, or with evidence on imaging or direct observation of lymph node metastases (recommendation # 35 ATA guidelines) [5].
The surgeon and pediatrician caring for the patient, in consultation with the child’s parents, should decide the timing of thyroidectomy. Children in the ATA-MOD category should have a history and physical examination, ultrasound of the neck, and measurement of serum calcitonin levels beginning around 5 years of age (recommendation # 36 ATA guidelines) [5]. The timing of thyroidectomy should be based on the detection of an elevated serum calcitonin levels; however, six month or annual evaluations may extend to several years or decades. Parents who are worried about a long-term surveillance program may elect to have their child’s thyroid gland removed around five years of age (recommendation # 36 ATA guidelines) [5].
The surgeon and pediatrician caring for the patient, in consultation with the child’s parents, should decide the timing of thyroidectomy.
In patients with hereditary MTC screening protocols for pheochromocytoma ought to start by age 11 years for children in the ATA-H and ATA-HST categories and by age 16 years in children in the ATA-MOD category (recommendation # 37 ATA guidelines) [5]. Screening entails determining free plasma metanephrines and normetanephrines or 24-hour urinary metanephrines and normetanephrines. Adrenal imaging with CT or MRI is indicated in patients with positive biochemical results (recommendation # 37 ATA guidelines) [5].
Patients with MEN2A or MEN2B and a histological diagnosis of MTC irrespective of age and presenting symptoms must have a pheochromocytoma rule out prior to any interventional procedure (recommendation # 38 ATA guidelines) [5].
A pheochromocytoma must be rule out in women of childbearing age with MEN2A or MEN2B who are planning a pregnancy or are pregnant (recommendation # 38 ATA guidelines) [5]. If a pheochromocytoma is diagnosed it should be treated preferably prior to the third trimester (recommendation # 38 ATA guidelines) [5]. If the pheochromocytoma coexists with either MTC of PHPT it should be removed prior to the operation for either MTC or PHPT (recommendation # 39 ATA guidelines) [5]. After a proper preoperative preparation, the pheochromocytoma should be removed via a laparoscopic or retroperitoneoscopic adrenalectomy (recommendation # 40 ATA guidelines) [5]. Subtotal adrenalectomy to preserve adrenal cortical function should be contemplated as an alternative operation (recommendation # 40 ATA guidelines) [5]. After and adrenalectomy the patients should be educated regarding the risk of adrenal crisis and wear a bracelet or a necklace indicating that they have no adrenal glands and are on corticosteroid replacement therapy. These patients require glucocorticoid and mineralocorticoid replacement therapy and should be carefully monitored to ensure that their steroid levels are adequate (recommendation # 41 ATA guidelines) [5]. Glucocorticoid replacement therapy will be needed if they become severely ill or are injured.
Patients in the ATA-H and ATA-MOD categories should be screened for PHPT at the time of screening for pheochromocytoma (by age 11 years in patients in the ATA-H category and by age 16 years in patients in the ATA-MOD category) (recommendation # 42 ATA guidelines) [5]. When operating on patients with PHPT only visibly enlarged parathyroid glands should be removed (recommendation # 43 ATA guidelines) [5].
If all four glands are enlarged management options include subtotal Para thyroidectomy or total Para thyroidectomy with auto transplantation (recommendation # 42 ATA guidelines) [5]. Patients who develop PHPT following a thyroidectomy for MTC should have localization studies performed prior to the operation (recommendation # 43 ATA guidelines) [5].
At reoperation all enlarged parathyroid glands should be removed, and parathyroid of normal size should be left in situ. If only one enlarged parathyroid gland is identified and there is histological documentation that three parathyroid glands have been removed previously, a portion of the enlarged gland should either be left in situ with an adequate blood supply or auto transplantation should be performed (recommendation # 43 ATA guidelines) [5].
2.8. Postoperative Management
Levothyroxine (thyroxine, T4) therapy should commence immediately after the operation; an appropriate initial dose is 1.3 to 1.6 mcg/kg of body weight [82,83].
The appropriateness of therapy should be assessed clinically and by obtaining serum thyroid-stimulating hormone (TSH) approximately four to six weeks after initiating replacement therapy (recommendation # 31 ATA guidelines) [5]. The goal of levothyroxine therapy should be to reestablish and maintain a thyroid state (recommendation # 31 ATA guidelines) [5]; suppression of serum TSH concentrations is not indicated in patients with MTC due to the fact that the perifollicular C cells are not responsive to TSH. Likewise, adjuvant therapy with radioiodine is contraindicated, because the MTC tumor cells do not concentrate iodine [84].
Total calcium serum levels should be monitored postoperatively. Patients who develop symptomatic hypocalcemia should receive oral calcium and vitamin D supplementation. Chronic replacement therapy is indicated in patients who cannot be weaned from medication (recommendation # 32 ATA guidelines) [5].
Somatic mutations in RET, HRAS, KRAS, or rarely, NRAS, can be identified in tumors of patients with sporadic MTC as discussed in previous sections. Some studies [23,73,76], but not all [77], have suggested that the identification of the RET mutation implies a more aggressive course than those without the mutation. Nevertheless, ATA does not recommend routine somatic mutational analysis of tumor samples [5].
After initial therapy, it is important to evaluate patients to determine if surgery was curative [85]. Physicians should consider TNM classification, the number of lymph node metastases, and postoperative calcitonin levels in predicting outcome and planning long-term follow-up of patients treated by thyroidectomy for MTC (recommendation # 45 ATA guidelines) [5]. Various associations recommend measuring serum calcitonin and CEA to assess for cure [5,6,47].
Subsequent management depends upon these values. Calcitonin and CEA levels should be obtained approximately three months after surgery to detect for the presence of residual disease and if undetectable or within the normal range, they should be measured every six months for one year, and then yearly thereafter (recommendation # 46 ATA guidelines) [5].
Patients who have normal serum CEA and untraceable calcitonin levels are deemed biochemically cured and have the best prognosis (five-year recurrence rate around 5%) [85-89]. Further imaging is not required unless the calcitonin or CEA values rise during follow-up. The calcitonin concentrations fall gradually overtime in some patients, with the nadir not being reached for several months so the timing of when to obtain these values is crucial (around two to three months after the operation) [90]. Nonetheless, in patients that are cured, calcitonin concentrations start to decline very fast within the first postoperative hour [90], often attaining undetectable levels within the first few postoperative days [91,92].
Patients with elevated postoperative calcitonin concentrations less than 150 pg/mL should have a physical examination and neck ultrasound. If these studies are negative the patients should be followed with physical examinations, serum calcitonin and CEA, and ultrasound every six months (recommendation # 47 ATA guidelines) [5].
If the postoperative calcitonin levels are found to be above 150 pg/mL patients should be evaluated by imaging procedures, including neck ultrasound, chest CT, contrast-enhanced MRI or three-phase contrast-enhanced CT of the liver, and bone scintigraphy and MRI of the pelvis and axial skeleton (recommendation # 48 ATA guidelines) [5].
The liver is the most common site of distant metastases in patients with MTC, occurring roughly in 45% of patients with advanced disease [5]. Other sites of distant metastasis include bone, brain, and lung. If imaging work-up is negative, monitoring with history and physical exam, calcitonin and CEA levels, and evaluation of the neck with ultrasonography should continue.
The frequency of repeating imaging studies will be dependent on the magnitude and rate of rise of the calcitonin and CEA levels. Patients with stable postoperative calcitonin levels in the 150 to 300 pg/mL range are usually followed with yearly neck ultrasound for several years, reserving repeat cross-sectional imaging (neck, chest, abdomen, pelvis) looking for distant metastases in those patients with rising calcitonin or CEA levels. PET/CT scans are considered only when the calcitonin concentrations are higher than 500 to 1000 pg/mL [93]. Radionuclide bone scan can be helpful in selected cases when cross-sectional imaging fails to identify the source of the elevated calcitonin levels [94-96].
Patients with measurable calcitonin and CEA levels following thyroidectomy, the levels of the markers should be measured at least every six months to determine their doubling times (recommendation # 49 ATA guidelines) [5]. An elevated basal calcitonin level three months after surgery is probable evidence of residual disease. Thirty to 55% of patients with palpable MTC or nonpalpable but macroscopic MTC who undergo attempted curative resection have persistently elevated calcitonin concentrations [46,89,97-99].
The prognosis for patients with postoperative elevations of calcitonin levels depends primarily upon the patient's age and the extent of disease at the time of initial surgery [45,46,89,100].
In a study of 899 patients with MTC the 10-year survival in the patients with elevated postoperative calcitonin levels was 70%, compared with 98% in patients who were biochemically cured [89]. Younger age at the time of the operation and no lymph node involvement were predictive of biochemical cure. Postoperative RAI is not indicated following thyroidectomy for MTC; however, it should be considered in patients whose regional or distant metastases contain MTC mixed with either Papillary Thyroid Carcinoma (PTC) or Follicular Thyroid Carcinoma (FTC) (recommendation # 52 ATA guidelines) [5].
Surgical resection of persistent or recurrent local and regional MTC in patients without distant metastases should include compartmental dissection of image-positive or biopsy-positive disease in the central (level VI) or lateral (levels II to V) neck compartments (recommendation # 50 ATA guidelines) [5]. Limited operative procedures, such as resection of only grossly metastatic lymph nodes, should be avoided unless there has been prior extensive surgery in a compartment (recommendation # 50 ATA guidelines) [5].
Adjuvant external beam radiation therapy (EBRT) to the neck and mediastinum should be considered in patients at high risk for local recurrence (microscopic or macroscopic residual MTC, extra thyroidal extension, or extensive lymph node metastases), and those at risk of airway obstruction (recommendation # 52 ATA guidelines) [5]. The potential benefits must be weighed against the acute and chronic toxicity linked with this therapy.
Systemic therapy should not be administered to patients who have increasing calcitonin and CEA levels but no documented metastatic disease. Systemic therapy should not be administered to patients with stable low-volume metastatic disease, as determined by imaging studies and calcitonin and CEA doubling times greater than 2 years (recommendation # 53 ATA guidelines) [5].
When suspecting persistent or recurrent MTC following thyroidectomy, one should contemplate laparoscopic or open evaluation and biopsy of the liver to exclude occult metastases before exposing them to a long and grueling repeat neck operation (recommendation # 54 ATA guidelines) [5].
Management options for patients with recurrent or residual disease include observation / active surveillance, surgical resection, EBRT, and directed therapies (such as radiofrequency ablation, cry ablation, embolization), or systemic therapies. At one point in time, patients with detectable residual or recurrent MTC were routinely operated on. Nonetheless, despite routine lymphadenectomy or excision of palpable tumor, the calcitonin concentrations often did not normalize after these operations [101].
This has led to a more critical appraisal for the need for and timing of therapeutic interventions and re-evaluation of the role of cautious observation in properly selected patients. The following approach to management of residual disease is based upon observational studies and clinical experience.
The suggested management algorithm for persistent or recurrent disease depends upon a variety of clinical factors, including: whether or not the disease can be localized, volume of disease, exact location of the metastatic disease, whether or not the disease is symptomatic, and the likelihood of progression of clinically significant structural disease.
Patients with detectable calcitonin and/or abnormal CEA levels without structurally identifiable disease are best followed with observation. The extent and frequency of cross-sectional imaging is dependent on the magnitude and doubling time of calcitonin and CEA levels. In the absence of structurally identifiable disease, the guidelines do not recommend additional surgery or systemic therapies [5,26,27,47].
In the absence of macroscopic (R2) residual disease, the guidelines not recommend routine use of postoperative EBRT as adjuvant therapy, even if the postoperative calcitonin and CEA levels are abnormal [5,26,27,47]. Adjuvant EBRT is reserved for very select patients with extensive preoperative macroscopic extra thyroidal extension or large volume, multicompetent macroscopic lymph node involvement [5].
The absence of randomized prospective trials makes it difficult to provide definitive recommendations with regard to the use of EBRT as adjuvant therapy. Some authors feel that EBRT in this setting may improve local and regional control but are not convinced that there is an overall survival benefit [102]. In a retrospective series of 51 patients with persistently elevated calcitonin levels (ranging from 540 to 400,000 pg/mL) and no evidence of residual disease, the local relapse rate was substantially lower in those who were treated with EBRT (29% versus 59%) [103]. The 10-year survival rate between the two groups was not statistically significant (72% versus 60%) [103].
Small volume disease outside of regional lymph nodes is usually the consequence of gross extra thyroidal extension into major structures in the neck, and it often indicates microscopic disease remaining after all macroscopic disease has been surgically removed (R1 resection). Typically, this is microscopic disease involving the soft tissues in the neck, the strap muscle, the trachea, esophagus, or thyroid bed. As these patients are at high risk of local and regional recurrence, EBRT is recommended as adjuvant therapy [5,26,27,47].
Structurally identifiable small volume disease can also be a manifestation of an early soft tissue recurrence of MTC, in which case the initial treatment option would be surgical resection, which is then usually followed by EBRT (or occasionally EBRT alone if surgery is unlikely to be successful or likely to result in unacceptable morbidity) [102]. Retrospective data have implied that EBRT may prolong the interval until disease progression or recurrence in some patients. Brierley et al. Reported 10-year local and regional control rates of 86% in patients with residual microscopic neck disease who received postoperative radiotherapy versus 52% in those who did not [102].
MTC patients with asymptomatic, persistent, small volume local and regional disease (sub centimeter lymph node metastases), active surveillance is the best management option. Surgical management is frequently not curative and can be associated with a high morbidity (hypoparathyroidism, injury to recurrent laryngeal nerve or accessory nerve). Furthermore, observation of stable, asymptomatic, lymph node metastases is almost always recommended if the basal serum calcitonin is greater than 1000 pg/mL or if more than five metastatic lymph nodes were removed with a previous surgery, as reoperation is almost never curative in these settings [5].
Patients are followed with serial cross-sectional imaging at six to 12-month intervals, and surgical intervention is reserved for patients that have documented structural disease progression [5]. Small volume distant metastases (sub centimeter metastatic foci) are usually followed with observation. It is not unusual for small pulmonary and liver metastases to continue asymptomatic and progress very slowly (or not all) over many years [5].
If the macroscopic residual disease is confirmed to be unrespectable or if the patient / multidisciplinary team agree that a complete surgical resection would produce unacceptable morbidity, EBRT may be utilized to improve local and regional control [5,47]. Another viable option is systemic therapy (such as Tyrosine Kinase Inhibitors [TKI]) [47].
Resection of large volume local and regional lymph node disease may be necessary to prevent invasion into surrounding major structures. Sporadically, lymph node metastases can manifest clinically as painful lesions that can be readily palliated with surgical resection (recommendation # 62 ATA guidelines) [5]. Conversely, additional surgery rarely achieves a biochemical cure if the basal calcitonin levels are greater than 1000 pg/mL or if more than five metastatic lymph nodes were removed with a previous surgery, careful observation or systemic therapies (TKI) can be considered for asymptomatic large volume lymph node metastases discovered in this setting [47].
Brain imaging should be obtained in patients with metastatic MTC and neurologic symptoms, including patients who are candidates for systemic therapy. Patients with isolated brain metastases are candidates for surgical resection or EBRT (recommendation # 55 ATA guidelines) [5]. MTC patients with spinal cord compression secondary to metastatic disease require urgent management with steroid therapy and surgical decompression. If patients are not candidates for surgery EBRT alone should be administered (recommendation # 56 ATA guidelines) [5].
Patients with pathologic fractures or impending fractures require treatment. Management options include surgery, thermoablation (radiofrequency or cryotherapy), cement injection, and EBRT (recommendation # 57 ATA guidelines) [5]. Treatment with denosumab or bisphosphonates is advocated for patients with painful osseous metastases (recommendation # 58 ATA guidelines) [5].
Resection should be contemplated in patients with large solitary pulmonary metastases. Radiofrequency ablation should be considered when the metastases are peripheral and small. Systemic therapy with TKI should be considered in patients with multiple metastases that are progressively increasing in size (recommendation # 59 ATA guidelines) [5]. Resection may be considered in patients with large isolated hepatic metastases (recommendation # 60 ATA guidelines) [5].
Chemoembolization should be considered in patients with disseminated tumors less than 3 cm in size involving less than a third of the liver (recommendation # 60 ATA guidelines) [5]. If possible, cutaneous metastases should be surgically removed (recommendation # 61 ATA guidelines) [5]. Multiple cutaneous lesions are best managed by EBRT or ethanol injection (recommendation # 61 ATA guidelines) [5].
Palliative therapy, including surgery, EBRT, or systemic therapy, should be considered in patients with metastases causing pain, mechanical compression, or signs and symptoms of hormonal excess (recommendation # 62 ATA guidelines) [5].
Diarrhea is typically associated with large volume liver metastases, and the diarrhea associated with MTC can have a major impact on quality of life. Dietary measures such as avoiding alcohol intake and maintaining a diet that limits high fiber foods can be improved with anti-motility agents (lope amide, diphenoxylate-atropine, or codeine) as first-line management. Somatostatin analogs may provide reasonable symptomatic improvement in some patients. In selected patients, debunking of large tumor deposits with surgery or chemoembolization may improve diarrhea (recommendation # 62 and 66 ATA guidelines) [5]. Vandetanib (prior to medical or surgical adrenalectomy) as first-line treatment option for ectopic Cushing's syndrome, based on recent reports of a very rapid decrease in cortisol levels in several patients [104,105]. Since Cushing's syndrome is linked with severe and debilitating adverse effects, management should be keenly contemplated even in the setting of large volume persistent / recurrent disease (recommendation # 67 ATA guidelines) [5].
The use of single agent or combination cytotoxic chemotherapeutic regimens should not be administered as first-line therapy in patients with persistent or recurrent MTC given the low response rates and the advent of promising new treatment options (recommendation # 63 ATA guidelines) [5].
Therapy with radiolabeled molecules or pre-targeted radio-immunotherapy may be contemplated in selected patients, ideally in the setting of a well-designed clinical trial (recommendation # 64 ATA guidelines) [5]. In patients with significant tumor burden and symptomatic or progressive metastatic disease according to RECIST treatment with TKI targeting both RET and VEGFR tyrosine kinases should be considered as systemic therapy. The TKIs Vandetanib or cabozantinib can be used as single-agent first-line systemic therapy in patients with advanced progressive MTC (recommendation # 65 ATA guidelines) [5,47].
In MTC the TKI function to stimulate tumor proliferation, angiogenesis, invasion, and metastasis. Small molecule inhibitors of select tyrosine kinases have been studied for the management of advanced MTC, given the oncogenic role of inherited and somatic mutations in the RET gene, as well as the related roles of tyrosine kinases in growth factor receptors such as the Vascular Endothelial Growth Factor Receptor (VEGFR) [106,107]. These TKI partially inhibit multiple kinases at Nano molar concentrations and often affect multiple signaling pathways [107].
The growing comprehension of the molecular oncogenesis in MTC has led to the detection of new molecular targets. Improved understanding of the molecular pathogenesis of MTC has led to the testing of targeted molecular therapies. Imitanib Mesylate (Gleevac) is a known tyrosine kinase inhibitor already in clinical use against chronic xylogenous leukemia and gastrointestinal stromal tumors, targeting specific tyrosine kinases. Studies performed in vitro of these drugs showed dose-dependent inhibition of MTC cells and inhibition of phosphorylation of the RET protein [108-110]. In a phase II trial, no remissions were noted in nine patients treated with Imitanib Mesylate [111].
In the United States cabozantinib and vandetanib are approved for management of symptomatic or progressive MTC in patients with unrespectable locally advanced or metastatic disease thanks to the results of phase III randomized studies that showed significant prolongation of progression-free survival and partial responses in approximately 20% to 50% of patients. Even though complete response rates are extremely rare, TKIs can possibly offer long-term disease stabilization. Nonetheless, data on the ability of any of these agents to improve survival are limited [106,112-115].
Vandetanib (ZD6474, Zactima) is a new oral anilinoquinazoline compound engineered to selectively inhibit Vascular Endothelial Growth Factor Receptor (VEGFR), Endothelial Growth Factor Receptor (EGFR), and RET tyrosine kinases [94]. Vandetanib does not work in all germline mutations of the RET gene, and thus its efficacy must be studied in patients with specific germline mutations identified [112,116]. In a phase II trial restricted to patients with measurable metastatic or unrespectable hereditary MTC, 30 patients received Vandetanib at a dose of 300 mg a day, until disease progression or adverse events occurred [113]. Primary end points were tumor response, with secondary end points being rate of disease control and biochemical response. A partial response was noted in 20% of patients, and 53% of patients had stable disease lasting at least 24 weeks [113].
Calcitonin levels showed 50% decrease from baseline that was maintained for at least 6 weeks [113]. The most common side effects that occurred in more than 50% of the patients were diarrhea, rash, fatigue, and nausea. In another study examining a lower-dose of Vandetanib (100 mg daily) in 19 patients with unrespectable or metastatic hereditary MTC, the response rates were very similar, partial responses were reported in 16% of patients and stable disease lasting at least 24 weeks in 53% patients [114]. Even though the dose was lower, side effects were very similar. During the study period, four patients with disease progression while taking the 100 mg dose received post progression treatment with 300 mg.
An international, randomized phase III trial of Vandetanib (300 mg daily) was performed in over 300 patients with unresectable locally advanced or metastatic sporadic or hereditary MTC. After a median follow-up of 24 months, progression-free survival was considerably prolonged for patients allocated to Vandetanib versus placebo (Hazard Ratio [HR] 0.46, 95% CI 0.31-0.69) [115]. The median progression-free survival for the Vandetanib group was predicted to be 30.5 months compared with 19.3 months in the placebo group. The objective response rate was considerably higher in the Vandetanib group (45% versus 13%). No difference has been detected in overall survival between the two treatment arms despite the improvement in progression-free survival. Patients with both progressive and stable disease were eligible for enrollment, and outcomes were similar in the two groups. Though, patients with CEA doubling times greater than 24 months were unlikely to benefit from treatment. The presence of a somatic RET M918T mutation predicted an improved progression-free survival [115]. Frequent side effects affecting ≥ 20% of patients include diarrhea/colitis, rash, dermatitis, nausea, hypertension, headache, fatigue, anorexia, abdominal pain, hypocalcemia, decreased glucose, and increased alanine aminotransferase (ALT) [115]. Severe adverse effects (occurring in ≥ 5% of the patients) included diarrhea/colitis, hypertension and hypertensive crisis, QT prolongation, fatigue, rash, torsades de pointes and sudden death.
Cabozantinib is an oral, small molecule TKI that targets VEGFRs 1 and 2, c-MET, and RET [117], that was approved by the US Food and Drug Administration (FDA) for the treatment of progressive, metastatic MTC [118]. In a phase I, dose-escalation study, 29% of patients with MTC achieved a partial response [117]. Stable disease of at least six months was observed in 41% of patients with MTC. The overall rate of partial responses and six-month, progression-free survival was 68% [117]. The drug is active in patients without RET activating mutations.
In the EXAM trial, 330 patients with progressive, metastatic or unrespectable locally advanced MTC were randomly assigned to receive either cabozantinib (140 mg) or placebo once daily [119]. Minimum follow-up was 42 months. Kaplan-Meier analysis showed a 5.5-month increase in median overall survival with Cabozantinib versus placebo (26.6 versus 21.1 months) although the difference did not reach statistical significance [stratified Hazard Ratio (HR), 0.85; 95% Confidence Interval (CI), 0.64-1.12; P = 0.24]. In an exploratory assessment of overall survival, progression-free survival, and objective response rate, cabozantinib appeared to have a larger treatment effect in patients with RET M918T mutation-positive tumors compared with patients not harboring this mutation. For patients with RET M918T-positive disease, median overall survival was 44.3 months for Cabozantinib versus 18.9 months for placebo [HR, 0.60; 95% CI, 0.38-0.94; P = 0.03 (not adjusted for multiple subgroup analyses)], with corresponding values of 20.2 versus 21.5 months (HR, 1.12; 95% CI, 0.70-1.82; P = 0.63) in the RET M918T-negative subgroup. Median treatment duration was 10.8 months with Cabozantinib and 3.4 months with placebo. Lower starting doses, such as 60 mg used for other malignancies, are also well tolerated but not formally examined yet for this tumor. It is preferable that patients with progressive advanced or symptomatic MTC participate in clinical trials of targeted therapies. On the other hand, for those patients who are unwilling or unable to participate in clinical trials, either Cabozantinib or Vandetanib is recommended per guidelines [5,47].
The oral, small molecule TKI, Sorafenib, targets the VEGFR 2 / 3 and most mutant forms of RET [120]. In a pilot study, patients with metastatic MTC (N=5) were treated with Sorafenib, starting at 400 mg twice daily [121]. After six months of treatment, responses were seen in two patients (including one complete response) and symptomatic improvement was seen in all, but most patients required a dose reduction due to side effects. Furthermore, preliminary data from a larger (n = 16), open-label, phase II study of Sorafenib in patients with metastatic MTC showed a partial response in one patient with sporadic MTC and a median progression-free survival of nearly 18 months [122]. Partial response (n = 3) or durable stable disease (n = 3) was also reported in six of eight MTC patients participating in a phase I study of combination sorafenib and tipifarnib [123]. Sorafenib is approved for the management of advanced renal cell carcinoma and unrespectable hepatocellular carcinoma and could be considered for use in very selected patients with advanced MTC who are unable to participate in clinical trials.
Preliminary data of an oral, small molecule TKI that targets all three VEGFRs and RET in MTC, called Sunitinib [124], shows promising results. In one study a patient experienced a prolonged partial response using the following regimen, 50 mg daily for 28 days followed by 14 days of no treatment per cycle [125]. In a phase II, open label trial, in patients with progressive refractory thyroid cancer (N=7 with MTC) with a median follow-up of 15.5 months, three MTC patients had a complete or partial response, and disease stable disease occurred in two [126]. The interim analysis from a second open-label, phase II trial reported partial responses or stable disease for greater than 12 weeks in three of eight MTC patients [127]. Sunitinib is approved for the management of advanced renal cell carcinoma and refractory gastrointestinal stromal tumors and could be considered for use in very selected patients with advanced MTC who are unable to participate in clinical trials.
Lenvatinib is a TKI that is taken orally that targets VEGFRs, RET, and Fibroblast Growth Factor Receptors (FGFR) 1 to 4. In a phase II trial (N = 59) of surgically unrespectable, progressive MTC treated with Lenvatinib, starting at 24 mg daily the best overall response rate, that was partial in all cases, was 35% (95% CI 24% to 49%) [128]. An additional 44% had stable disease. Indistinguishable response rates were observed in the cohorts previously treated and never treated with prior VEGFR-targeted therapies. Median progression-free survival and overall survival were 9.0 months and 16.6 months, respectively [128]. Predictable side effects were observed, including diarrhea, hypertension, and decreased appetite. Based on this data the FDA in the United States approved Lenvatinib for the treatment progressive, metastatic, radioiodine-refractory differentiated thyroid cancer. Lenvatinib may be considered as a second- or third-line TKI therapy for patients with progressive, metastatic MTC who have failed other anti-VEGFR therapies.
Pazopanib is a potent, orally given, small molecule TKI that targets all VEGFR subtypes but has no significant inhibitory activity against RET [129]. Hence, its actions are expected to be primarily antiangiogenic in MTC. In a study on 35 patients with rapidly progressing MTC treated in a phase II trial, 14% of patients experienced a partial response, lasting 29 weeks [130]. Twenty-three percent were alive without progression, 14% were alive with progression, 63% had died [130]. The median progression-free was estimated to be 9.4 months and the overall survival times was estimated to be 19.9 months. The efficacy of pazopanib to induce objective responses in the absence of significant anti-RET activity suggests that RET may not be as important a target for therapy as the VEGFR. Pazopanib is approved in the United States for the treatment of advanced renal cell carcinoma and in the absence of more extensive data in MTC patients, it is not recommended for use in these patients.
Multiple other TKIs have been studied in clinical trials during the past several years, but these drugs remain investigational at this time and are not available for routine clinical use. Overall, the results of these studies are consistent with the findings described above, supporting the concept that antiangiogenic TKIs are useful treatments for advanced metastatic MTC. Newer agents likely to be evaluated in phase I trials in the near future will include kinase inhibitors that are highly selective for RET kinase with negligible activity against VEGFRs. Much work is still to be done to answer the question of how effective chemotherapy is at treating locally advanced or metastatic MTC. At best, many of these molecular chemotherapeutic targets appear to have a cytostatic effect on tumor progression, which, for a tumor that is as indolent and slow growing as MTC, could have significant clinical implications for this patient population.
MTC has biosynthetic activity that results in the production of CEA [58]. Molecularly targeted antibodies toward CEA have been used with limited efficacy in clinical trials. A study with the humanized anti-CEA monoclonal antibody Labetuzumab showed significant inhibition of MTC tumor growth in vivo, with sensitization of the tumors to conventional chemotherapy [108]. A study using the humanized anti-CEA monoclonal antibody Labetuzumab showed significant inhibition of MTC tumor growth in vivo, but in a phase I trial employing Labetuzumab there was only limited benefit in patients with advanced MTC [131]. The lack of a significant treatment response is thought to be related to the relationship between pharmacokinetics and tumor burden suggesting that the drug would likely be more successful in patients with early stage disease.
2.9. Prognosis
Age and stage of disease at the time of presentation has been shown to be an important factor that influences prognosis [5,46]. Five and 10-year Disease-Free Survival (DFS) rates are higher among patients 40 years old or less as compared with patients over age 40 years (95% versus 65% and 75% versus 50%, respectively) [44,46]. On the other hand, a different study found no effect of age if survival was compared with the expected mortality rates in the general population [132]. The 10-year survival rates for patients with stages I, II, III, and IV MTC are 100%, 93%, 71%, and 21%, respectively [5,89]. Controlling for the effect of age, the prognosis of patients with familial MTC is possibly similar to those with sporadic MTC [133,134]. Certain germline mutations in the RET gene predict the aggressiveness of the tumor [133,134].
Calcitonin and CEA doubling times provide sensitive markers for progression and aggressiveness of metastatic MTC. Calcitonin doubling times less than six to 12 months are associated with poor survival, while doubling times greater than 24 months are associated with a very favorable prognosis [70,71]. Additional factors that may predict a poor prognosis include paucity of tumor immunostaining for calcitonin [135], cellular heterogeneity, prominent tissue immunostaining for galectin-3 [136] or immunostaining for CEA associated with scant or absent tissue staining for calcitonin [137], high preoperative serum CEA [49], an elevated procalcitonin: calcitonin ratio [138-154], and a rising CEA level associated with a stable or declining calcitonin level.
3. Conclusion
MTC is a rare malignancy. Surgery remains the first-line treatment for any patient presenting with respectable MTC. The proclivity of MTC to metastasize to regional lymph nodes in the neck is a defining characteristic of this disease, which apprises surgical treatment in all clinical settings. Elimination of involved nodes can result in long-term cure or disease control, and a working knowledge of cervical lymph node anatomy and of the natural history of MTC spread within these nodal groups is important to the surgeon managing these patients. The parathyroid glands are closely related to the central compartment lymph nodes. Meticulous, management of the parathyroid glands in these situations minimizes the risk of hypoparathyroidism. Additional studies are needed to elucidate the role of EBRT in the armamentarium for local control post resection, advanced-stage MTC, and palliation. Ample research is still needed to find efficacious therapies for the treatment of persistent and recurrent unrespectable disease. In general, long-term survival in MTC patients remains high when managed in a multidisciplinary fashion following evidenced-based medicine.

Figure
1:
Single circumscribed but non-encapsulated, gray-tan tumor.

Figure 2: MTC stained with
hematoxylin and eosin.

Figure 3: Background of
amyloid.
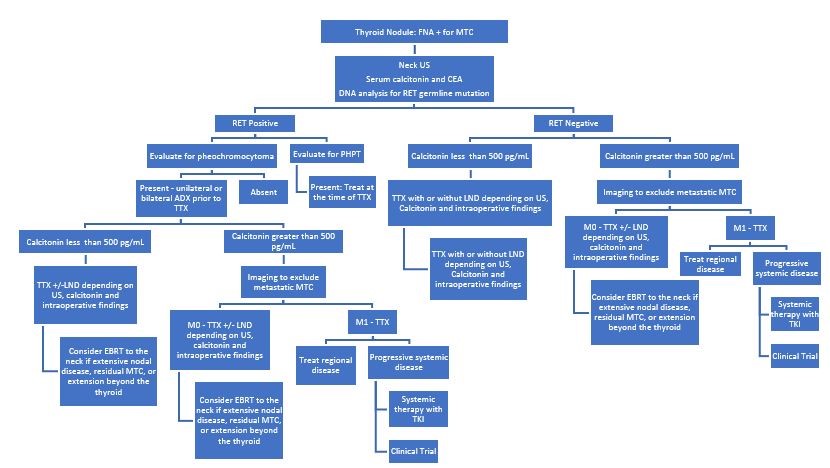
Figure 4: Management of
patients with a thyroid nodule and histological diagnosis of MTC.
|
Syndrome Manifestation |
Percentage (%) |
|
Classic MEN 2A:
|
100% 50% 10% to 20% |
|
Variants of MEN 2A:
|
|
|
MEN 2B:
|
100% 50% 0% > 90% > 90% |
Table 1: Clinical features of MEN type 2 syndromes.
|
Histologic Variants |
Nest-like pattern with pigmented dendritic cells resembling sustentacular cells [151]
|
Table 2: Variants of MTC based on the dominant histologic pattern.
|
T Category and Criteria |
|
TX - Primary tumor cannot be assessed T0 - No evidence of primary tumor T1 - Tumor ≤ 2 cm in greatest dimension limited to the thyroid T1a - Tumor ≤ 1 cm in greatest dimension limited to the thyroid T1b - Tumor > 1 cm but ≤ 2 cm in greatest dimension limited to the thyroid T2 - Tumor > 2 cm but ≤ 4 cm in greatest dimension limited to the thyroid T3 - Tumor > 4 cm or with extra thyroidal extension Tumor > 4 cm in greatest dimension limited to the thyroid Tumor of any size with gross extra thyroidal extension invading only strap muscles (stern hyoid, stern thyroid, thyrohyoid or omohyoid muscles) T4 - Advanced disease T4a - Moderately advanced disease; tumor of any size with gross extra thyroidal extension into the nearby tissues of the neck, including subcutaneous soft tissue, larynx, trachea, esophagus, or recurrent laryngeal nerve T4b - Very advanced disease; tumor of any size with extension toward the spine or into nearby large blood vessels, invading the prevertebral fascia, or encasing the carotid artery or mediastinal vessels |
|
N Category and Criteria |
|
NX - Regional lymph nodes cannot be assessed N0 - No evidence of loco regional lymph node metastasis N0a - One or more cytological or histologically confirmed benign lymph nodes N0b - Radiologic or clinical evidence of loco regional lymph node metastasis N1 - Metastasis to regional nodes N1a - Metastasis to level VI or VII (pretracheal, pretracheal, or prelaryngeal/Delphian, or upper mediastinal) lymph nodes. This can be unilateral or bilateral disease. N1b - Metastasis to unilateral, bilateral, or contralateral lateral neck lymph nodes levels I, II, III, IV, or V) or retropharyngeal lymph nodes |
|
M Category and Criteria |
|
M0 - No distant metastasis M1 - Distant metastasis |
Table 3: Definitions of the AJCC TNM Staging System [79].
|
T |
N |
M |
Stage |
|
T1 T2 T3 T1 - T3 T4a T1 – T3 T4b Any T |
N0 N0 N0 N1a Any N N1b Any N Any N |
M0 M0 M0 M0 M0 M0 M0 M1 |
I II II III IVA IVA IVB IVC |
Table 4: AJCC Prognostic Stage Groups [79]
3. Williams ED (1966) Histogenesis of medullary carcinoma of the thyroid. J Clin Pathol 19: 114-118.
6. Howlader N, Krapcho M, et al. (2017) SEER Cancer Statistics Review, 1975-2014, based on November 2016 SEER data submission, posted to the SEER web site. N C Institute Editor Bethesda MD.
7. Ryan Thomas M, Perrier, Elizabeth G. Grubbs (2012) Well Differentiated Carcinoma of the Thyroid and Neoplasms of the Parathyroid Glands. Fourth Edition ed The M D Anderson Surgical Oncology Handbook: Fifth Edition. Philadelphia, USA: Lippincott Williams and Wilkins. 1: 900.
8. Grubbs K (2018) Carcinoma of the Thyroid Gland and Neoplasms of the Parathyroid Glands. Sixth edition ed. The MD Anderson Surgical Oncology Handbook, ed. M. Barry W. Feig. 2019 Philadelphia, PA: Wolters Kluwer.
27. American Thyroid Association Guidelines Task F, et al., Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 19: 565-612.
29. Burk W (1901) Über einen Amyloidtumor mit Metastasen, in Pietzcker Inaugural-Dissertation. Tübingen Germany.
30. Stoffel E (1910) Lokales Amyloid der Schilddrüse. Virchows Arch 201: 245-252
31. Gagel RF, H A, Cote GJ (2005) Medullary thyroid carcinoma. 9th ed. The Thyroid, ed. U.R.B. LE. 2005, Philadelphia 2005: Lippincott Williams & Wilkins.
34. Wermer P (1954) Genetic aspects of adenomatosis of endocrine glands. Am J Med 16: 363-371.
35. Cancer of the Thyroid Invasive: Trends in SEER In- cidence and U.S. Mortality Using the Joinpoint Regres- sion Program, 1975-2011(SEER) Stat version 8.1.2 Rate Session. (Access the SEER 18 database at www.seer.cancer.gov). Incidence - SEER 18 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases, Nov 2012 Sub (2000-2010) < Katrina/Rita Population Adjustment > - Linked to County Attributes - Total U.S., 1969-2011 Counties, National Cancer Institute, DCCPS, Sur- veillance Research Program, Surveillance Systems Branch, released April 2013, based on the November 2012 submission.
47. NCCN (2017) NCCN Clinical practice guidelines in oncology: thyroid carcinoma 2.
60. LiVolsi VA (1997) C cell hyperplasia/neoplasia. J Clin Endocrinol Metab 82: 39-41.
72. Moline J, Eng C (2011) Multiple endocrine neoplasia type 2: an overview. Genet Med 13: 755-764.
79. Rosen JE, Brierley JD, Grogan RH, Haddad R, Hunt JL, et al. Thyroid - Medullary. American Joint Committee on Cancer Staging Manuel ed. M.B. Amin. Vol. 1. Springer International Publishing.
118. Cancer., U.F.a.D.A.F.a.C.t.t.r.t.o.t., Cabozantinib (CABOMETYX), U.F.a.D. Administration., Editor. 2016.
144. Beskid M, Lorenc R, Rościszewska A (1971) C-cell thyroid adenoma in man. J Pathol 103: 1-4.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.