Malignant Triton Tumor: A Narrative Review of Current Evidence and a Single Center Experience
by Irene Lanzetta1,2, Paolo Pedrazzoli1,2*, Francesca Rifaldi1,2, Silvio Sporeni1,2, Simone Figini1,2, Niccolò Leandro Alessio1,2, Anna Maria Clelia Gallotti3, Frediano Socrate Inzani4, Simona Secondino1,2
1Department of Internal Medicine and Medical Therapy, University of Pavia, Pavia, Italy
2Department of Oncology, IRCCS Policlinico San Matteo Foundation, Pavia, Italy
3Department of Radiology, IRCCS Policlinico San Matteo Foundation, Pavia, Italy
4Department of Pathology, IRCCS Policlinico San Matteo Foundation, Pavia, Italy
*Corresponding author: Paolo Pedrazzoli, Department of Oncology, IRCCS Policlinico San Matteo Foundation, Pavia, Italy
Received Date: 03 July 2024
Accepted Date: 08 July 2024
Published Date: 10 July 2024
Citation: Lanzetta I, Pedrazzoli P, Rifaldi F, Sporeni S, Figini S, et al (2024) Malignant Triton Tumor: A Narrative Review of Current Evidence and a Single Center Experience. Ann Case Report. 9: 1878. https://doi.org/10.29011/2574-7754.101878
Abstract
Background: Malignant Triton Tumor (MTT) is a rare type of neoplasm enclosed within the category of malignant peripheral nerve sheath tumors (MPNSTs). The pathogenesis is still unknown, even though it is frequently neurofibromatosis type 1-related. Diagnosis mainly relies on histological and immunohistochemical while the genetic investigation, is not diagnostic itself. Surgery, radiotherapy (RT), and systemic chemotherapy (CT) are employed treatment options.
Methods: Two cases of MTT diagnosed and treated at our institution are described. Furthermore, a web-based search of MEDLINE/PubMed library data published from 1998 to 2024 of English-language papers was performed to identify pertinent articles concerning MTT.
Results: Our study confirms that pathogenic and genetic paths remain unknown in MTT. The disease follows an aggressive course and treatment options still rely on local therapies, whenever feasible, and conventional CT. The only curative therapeutic option is the radical surgical excision, even though recurrence after excision is common and greater than that in MPNSTs. Adjuvant RT and CT can be used to obtain local control and prevent systemic relapse. In the advanced tumor setting a Doxorubicin and Ifosfamide combination is usually preferred, while subsequent therapeutic options remain uncertain. Our two patients received first-line treatment with Doxorubicin and Ifosfamide achieving a good disease response; one with locally advanced disease was then treated with surgery and RT.
Conclusions: Further evidence is needed to better define the most appropriate diagnostic and therapeutic pathways for these tumors. Patients with MTT should be referred to sarcoma centers and, whenever possible, accrued in phase I-II clinical trials.
Keywords: Malignant Triton Tumor; Malignant Peripheral Nerve Sheath Tumor; Treatment; Review.
Introduction
Malignant Triton tumor (MTT) is a rare disease that is commonly and academically enclosed within the category of malignant peripheral nerve sheath tumors (MPNSTs), from which it differs through a peculiar and unique composition consisting of both neoplastic Schwann cells and rhabdomyoblasts. MTT is named after the triton salamander1, which can develop supernumerary limbs formed by neurogenic and muscular components. The hystopathogenesis of MTT is still unknown; it has been hypothesized that multipotent neural crest cells of mesenchymal origin may differentiate into both muscle and nerve components, or that malignant Schwann cells can differentiate into rhabdomyoblasts [1,2]. MPNSTs represent 5 to 7% of soft tissue sarcomas (MTTs account for 5–10% of MPNSTs1) and are currently known to arise from Schwann cells, their precursors, or pre-existing neurofibromas3. In up to 50-70% of cases, MTT is associated with type 1 neurofibromatosis (NF1) [2]. For this reason, the disease’s onset usually occurs at a younger age (28-36 years) [4,5] than in sporadic cases (40-50 years).
MTT usually has a worse prognosis than MPNSTs, partly due to the difficulty in reaching the correct diagnosis and the possible treatment delay that this may entail. The 5-year overall survival (OS) rate of MTT stands around 5-20%, compared to 50-60% for MPNSTs in general [3,4]. This review aims to summarize the epidemiology, pathogenetic, and histologic features, provide the best available clinical data about MTT, and discuss the available treatments and outcomes of patients with this rare disease.
Methods
Taking advantage of the two cases of MTT diagnosed at our Institution, whose clinical history is reported separately, a webbased search of MEDLINE/PubMed library data published from 1998 to 2024 of English-language papers was performed to identify pertinent articles by using “malignant triton tumor” and “malignant peripheral nerve sheath tumors AND rhabdomyosarcoma”.A manual screening for references from original articles was also done to identify additional studies. We selected 29 articles. Only articles published in English were reviewed.
Pathogenesis
The pathogenesis of MTT has not yet been clarified. A few cases have been reported in literature likely radio-induced, with various latency times, both from radiation treatment (up to 8% of cases) or accidental exposure to high doses (some cases were indeed related to the Chernobyl disaster) [6]. However, the most meaningful cases of MTT, in which a pathogenic path could be identified, are related to type 1 neurofibromatosis (NF1), which suggests a key role in genetics. We can consider two types of tumors in the context of MTT, those associated with NF1 and those of sporadic onset. About 50-70% of MTT cases are estimated to be associated with NF11, and they occur mainly in the male sex at a young age (28-36 years). In this setting, tumors can develop after a long latency time of up to 10-20 years [7]. In contrast, those of sporadic onset are more associated with the female sex and older age (4044 years) [5] and may enter in differential diagnosis with other malignant spindle cells tumors such as rhabdomyosarcoma, fibro sarcoma, and malignant fibrous histiocytoma [7,8]. In addition, the localization site may also vary depending on the correlation to NF1: disease-related cases occur more frequently in the head and neck area, while others involve the trunk. However, there is still no evidence of the prognostic impact of this correlation; some authors argue that the coexistence of NF1 results in a worse prognosis with a greater tendency to develop metastasis, while others claim that it does not condition the outcome1 (Table 1).Considering the cases NF1-related, the most identified cytogenetic abnormality is the short-arm’s deletion of chromosome 17 (17p) [5]. The most reliable hypothesis is that the target involved in this deletion is the gene p53, to which the high-grade forms of sarcomas are linked. Few studies to date have examined the cytogenetic features concerning the cases of sporadic onset. There are no currently known cytogenetic abnormalities or specific chromosomal aberrations associated with the genesis of these tumors, however, most of these entities have shown complex karyotypes with multiple structural abnormalities. Some studies have identified recurrent breakpoints in MPNSTs: 7p22, 11q13-23, 20q13 and 22q11-135.
A breakpoint of interest involves the region 11p15, namely the region of myogenic differentiation, which is probably responsible for rhabdomyoblastic differentiation [7]. It is also reported in the literature that more than half of MTTs is related to abnormalities involving chromosome 1, causing breakpoints among multiple loci all along its length [9,10]. Another recurrent event that has been observed is the amplification of the c-myc oncogene [4,7], which is related to a more aggressive behaviour of the tumor. Also, some authors reported that aberrations in the hedgehog pathway were noted in MTT [4]. However, these few cytogenetic analyses do not allow us to reach a full overview of the possible role of genetics in the pathogenesis of MTT. Further studies will inevitably be necessary for the global understanding of the pathophysiology of these rare tumors.
Diagnosis
As proposed by Woodruff et al. [11], three criteria define the MTT: (1) the tumor arises along a peripheral nerve or in pre-existing type 1 neurofibromatosis or represents a metastasis from such a tumor; (2) the growth characteristics are similar to a Schwann cell tumor; (3) rhabdomyoblasts can be demonstrated within the tumor, and could not be attributed to either an extension or a metastasis derived from an extrinsic rhabdomyosarcoma.
Triton tumors can potentially affect any anatomical site. The most common ones are the proximal extremities and the trunk, and tumors that arise in non-extremity sites tend to have a worse prognosis. Also frequently, the head and neck region can be affected. Some cases of peculiar localization are reported in the literature, including orbit [12], trigeminal nerve [3], nasal cavity [13], brain, retroperitoneum, mediastinum, liver [14]; it also has been reported a case of MTT occurred on a vascularized free flap reconstruction graft [15].
Clinical Presentation
The diagnosis of MTT can sometimes be insidious, as the clinical onset may be nonspecific with mild symptoms or completely asymptomatic. Therefore, the disease can often be diagnosed in a late stage. Patients can present with a pronounced, rapidly growing superficial mass of firm texture on the skin surface, rarely associated with necrotic or hemorrhagic phenomena. Tumor growth could occur along the nerves involved and among the nearby soft tissues. In some cases, patients complain of sub-continuous pain or vague neurological symptoms; however, no cases of frank sensory or motor nervous deficits or severe neurological abnormalities are recounted in the literature. A personal or family history of NF1 should always make us consider MTT a possibility in the differential diagnosis.
Radiological features
Due to the scarcity of these tumors, radiological suspicion of MTTs is commonly difficult: imaging tends to be non-specific, roughly describing ill-defined margins, intratumoral lobulation, edema, calcification, and adjacent tissue destruction [16]. A computed tomography scan (CTS) seems to be more accurate than MRI, as it can show the lobulated shape of the mass, the hemorrhage, necrosis, and calcifications.
Histology and immunohistochemical features
Since MPNSTs are rare tumors, lacking strict histologic criteria, they are counted among the most challenging neoplasms to diagnose. Macroscopically, MTTs (and MPNSTs) appear as firm, pseudo-encapsulated masses with ill-defined margins and occasional phenomena of hemorrhage or necrosis5, especially in high-grade tumors. Microscopically, it is currently well established that MTT is composed of an MPNST-typical stroma (fasciculated pattern with spindle elements, which is also commonly found in fibrosarcomas, leiomyosarcomas, and synovial sarcomas) within rhabdomyoblasts that may be in a more or less abundant proportion and distribution. Rhabdomyoblasts usually appear as round cells with eosinophilic cytoplasm [5]. A pluridirectional differentiation may occur (about 15% of cases5), such as in additional mesenchymal or epithelial areas. The immunohistochemical features may demonstrate widespread or zonal positivity for desmin, musclespecific actin, and myogenin; S100; CK8/18; CD99, CD56, CD34; MYOD1; cytokeratin AE1/AE3; H-caldesmon. In greater detail, S100 protein focal positivity is recounted in 50 to 90% of MPNSTs [5] and enters in differential diagnosis with cellular schwannoma, in which the positivity is usually diffuse. Although few studies investigating the molecular pattern regarding MPNSTs demonstrated a high degree of genomic instability, and mutations in various genes such as NF1, CDKN2A/B, SUZ12, EED, TP53, and PRC2 inactivation, none of these features can be currently used to achieve a diagnosis [17].
Treatment options
Management strategies towards malignant MTTs are difficult to establish because of the rarity of the disease, and the definition of a treatment path is mainly based on the experience of individual centers or on data developed from retrospective studies [18].
Radical surgical excision is the mainstay of treatment and the only current therapeutic method with curative intent. Surgical management of MTT follows the common strategies used in other types of sarcomas: the main focus is the complete removal of the tumor, with histologically clear margins of resection [19]. Several studies have underlined the importance of surgical margin status and the complete surgical excision to achieve optimal local control and OS [20-23]. However, in some cases, wide en-bloc resection of major nerves is required with a subsequent potentially significant functional loss [24].
In conjunction with surgical resection, adjuvant or neoadjuvant radiotherapy (RT) can be performed to improve local control; in the neoadjuvant setting, RT is usually recommended for both intermediate and high-grade lesions, while concerning low-grade tumors it is recommended exclusively after a radical excision19. In a systematic review, McConnel et al. concluded that in the adjuvant setting a median dose of 50 Gy (range 4-66 Gy) over 30 fractions can be administered [25].
On the other hand, the role of systemic chemotherapy (CT) in MTT remains controversial, and much of the published data come from single institutional retrospective analyses. In literature, personalized adjuvant treatments that involve the concomitant use of Cisplatin and Paclitaxel are also reported [26].
Currently, the first-line CT regimen for large, unresectable, or metastatic tumor consists of a combination of Doxorubicin and Ifosfamide; this regimen is based upon data demonstrating activity in single agent and combination regimens utilizing anthracyclines and alkylators in soft tissue sarcomas [27]. Single agents that proved some activity against MTT are gemcitabine, docetaxel, carboplatin, etoposide, dactinomycin, vincristine, cyclophosphamide, and dacarbazine, but the clinical benefits with each of them are variable [26]. The second-line treatment remains uncertain, with some promising results being reported in Gemcitabine/Docetaxel or Carboplatin/Etoposide [19] regimens.
Case presentation
Two patients were closely observed in our hospital and diagnosed as MTT.
Case 1
A 58-year-old man, without any significant medical history, noted a superficial swelling located in the left scapular area in August 2023: he performed an ultrasound and subsequently an MRI, which led to the identification of a tumor of mesenchymal origin, dislocating the trapezius and other large regional muscles, of approximately 100x60x100mm, without any infiltration of the surrounding structures (Figure 1). A percutaneous ultrasoundguided histological sampling showed a predominantly spindle cell sarcoma, characterized at immunohistochemistry by focal expression of CKAE/AE3, S-100, CD99, CD56, MYOD-1 consistent proliferative index by Mib1/Ki67 of 30%; CD34, desmin, smooth muscle actin, caldesmon, EMA was negative. These features suggest the diagnostic hypothesis MTT (Figure 2).A staging CTS was performed with no evidence of distant disease.
After a multidisciplinary evaluation, the patient received a neoadjuvant treatment including CT and RT. The treatment included Doxorubicin (75 mg/m2 d 1-3 q 21) plus Ifosfamide (3000 mg/m2 d1-3 q 21), associated with primary prophylaxis with G-CSF. RT (50 Gy in 25 fractions) was performed after the second cycle. The treatment was well tolerated, without any significant side effects. Upon clinical re-evaluation showing disease response highlighted with a parametric reduction in size, the patient underwent radical surgery with the evidence (at our last visit in March 2024) of the absence of surgical complications and clinical or instrumental signs of disease recurrence.
Considering the limited solid data reported in the literature supporting a systemic CT treatment in addition to the radical surgical approach (as reported), adjuvant CT was not taken into consideration and the patient continued with his regular follow-up.
Case 2
A 58-year-old man, with a medical history of NF-1, hypokinetic dilated heart disease, and paroxysmal-persistent atrial fibrillation, underwent a CTS of the neck due to an increasing size of swelling in the left parotid region: the CT scan was performed in July 2023 and identified a mass with inhomogeneous contrast enhancement in the left parapharyngeal region of approximately 72x50x50mm, involving the deep lobe of the parotid, and imprinting the oropharynx; ipsilateral cervical nodes were also involved (Figure 3). Due to the subsequent onset of worsening dysphagia and rhinolalia, the patient underwent tracheostomy and PEG packaging and began enteral nutrition. A biopsy of the parapharyngeal mass was performed; histological examination revealed a solid proliferation composed mostly of spindle-shaped cells, with severe nuclear atypia, amidst significant rhabdomyoblastic component. Neoplasm showed, at immunohistochemistry, zonal positivity for S-100 and, in the rhabdomyoblastic component, expression of desmina, myogenin, and focally for MYOD1; MDM2, caldesmon, SOX-10, and GFAP resulted negative. Histological features, considering also the medical history of type 1 neurofibromatosis, resulted consistent with the diagnosis of MTT (Figure 4).
A staging CTS was then performed showing a vascular infiltration of the disease, an extensive thrombosis of the internal jugular vein and multiple bilateral lung metastases. According to the performance status of the patient, first-line CT was started with Doxorubicin (60 mg/m2 d 1-3 q 21) plus Ifosfamide (3000 mg/ m2 d 1-3 q 21), associated with primary prophylaxis with G-CSF. Despite the dose reduction, the first cycle was complicated by grade 4 febrile neutropenia and grade 4 thrombocytopenia, requiring hospitalization. Antibiotic therapy with Piperacillin/ Tazobactam was carried out until the recovery from hematological toxicity. From the second cycle on, due to the previous toxicities, the dosage of Ifosfamide was reduced by 20%, with consequent good tolerance, without significant side effects.
Upon clinical and instrumental re-evaluation, an initial response to treatment was observed, with resolution of pain and dimensional reduction of both the parapharyngeal mass and the lung metastases. After 6 cycles of CT, the patient had a very good partial remission of up to 90% of the lung metastases, and of the primitive tumor up to 50%, and He is going to receive local RT.
Discussion
MTTs are highly malignant tumors that represent 5-10% of all MPNSTs [1]. Almost 60% of cases are associated with NF1, while the remaining 40% seem to have a sporadic presentation, of which no pathogenetic cause has yet been identified, except for the correlation with radiation exposure [5].
In our report, case 1 displayed a sporadic presentation of the disease with histological features of S100-, desmin-, actin-, CD34- and MIB1/Ki67 proliferation index of 20-30% while case 2 displayed a typical association with NF1, with S-100+ (as in up to 90% of MTTs), desmin+, actin+, myogenin+, CK8/18+, MYOD1+, with an onset age distant from the median of such typical cases (2836 years) [5].As proposed by Woodruff et al. in 197311, which still dictates the inclusion criteria for a malignant Triton tumor’s diagnosis, both our cases present all the histopathological criteria required.
Regarding the histopathogenesis of this tumor, the most established theory is that multipotent neural crest cells of ectomesenchymal origin are capable of divergent differentiation to both nerve and muscle components1. Alternatively, Kamperis et al. demonstrated in a rat-model experiment that neoplastic Schwann cells possess the ability of mesenchymal differentiation into rhabdomyoblasts under stimulation by motor nerves [28].
The mechanism of presentation for Triton tumors remains elusive, but they typically present as a rapidly enlarging mass with associated pain and compressive symptoms to adjacent organs and tissues. However, their association with nerves or nervous lesions is well noted. In the early stage, patients often have no self-reported symptoms, and only seek medical attention when pain or dysfunction occur, as occurred for both of our patients. The literature search showed that in the majority of cases nerves including the cervical and brachial plexus, optic nerve, sciatic nerve, cervical sympathetic nerve, and spinal nerve root are involved [4]. There was no conclusive evidence that the masses in our cases came from such nerves; however, the onset localizations were observed in or around the parapharyngeal and paravertebral spaces.
As reported, CTS is the preferred instrumental strategy to identify this type of lesions, since is capable of visualizing bone erosion and providing indications of its malignancy and extension such as the lobulated shape of the mass, the hemorrhage, necrosis, and calcifications.
Nevertheless, MRI should be taken in consideration by the fact the origin of this type of tumors is from soft tissues. In certain images, ring or linear strips were observed, likely due to necrosis and hemorrhage in the mass as a result of quick growth. Also, 18FDGPET-CT is considered to be an effective method of estimating the disease spread [1]. One major area of concern for clinicians in MTT patients is the rate of tumor recurrence and relapse. Literature suggests that the local recurrence rate varies from 22% to 43% and metastases have been reported in 20% to 50% of cases1, with a 5-year survival rate of around 5–20% [3,4]. Recurrence depends on several factors, including tumor location, the extent of surgical excision, degree of differentiation, growth pattern, performance status, and comorbidities3. The most common metastatic sites are lungs, but other common extrapulmonary sites include the central nervous system, bone, liver, and the peritoneum [29].
Currently, there is no consensus regarding the treatment guidelines specific to MTTs. Nevertheless, as aforementioned, the only curative treatment is the radical surgical excision, while neoadjuvant or adjuvant radiotherapy and chemotherapy are given when the disease’s extension doesn’t allow a primary surgical approach, or to achieve local and systemic control. Indeed, previous experiences demonstrated that complete resection is an independent prognostic factor, and it decreased the mortality risk, prolonging postoperative survival [4]. Currently, the preferred first-line CT regimen consists of a combination of Ifosfamide and Doxorubicin [27]. Due to the limited availability of secondline therapies patients should be enrolled on ongoing phase I or phase II studies (Table 2) or undergo extensive molecular profiling and discussion within molecular tumor boards to identify if any therapy based on genomic alterations is available.
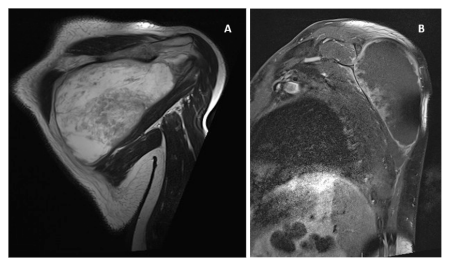
Figure 1: Coronal MRI T2-weighted image shows large expansive mass, hyperintense in T2w in the scapular region (A); Para-sagittal MRI T1-weighted imaging after MdC: the mass shows only peripheral irregular enhancement (B).
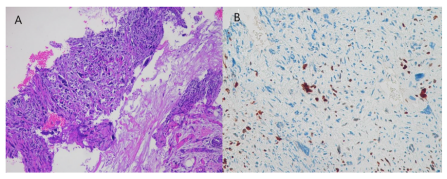
Figure 2: US-guided bioptic samples of scapular lesion consist, at histological examination, in minute fragments of a prolifefation composed by splindle atypical cells (A), and focally, some elements with rhabdomyoblastic differentiation as suggested by expression of Myod-1 (B). H&E (A), immunoperoxidase (B).
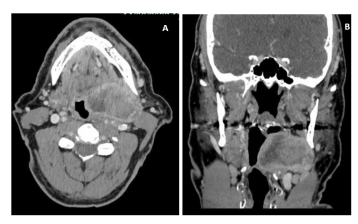
Figure 3: Axial CT after contrast medium: a large mass in the left parapharingeal space, with peripheral enhacement. The carotid vessels and jugular vein are posteriorly located (A). Coronal CT after contrast medium: the laterocervical mass imprints the left lateral wall of the oropharingeal cavity (B).
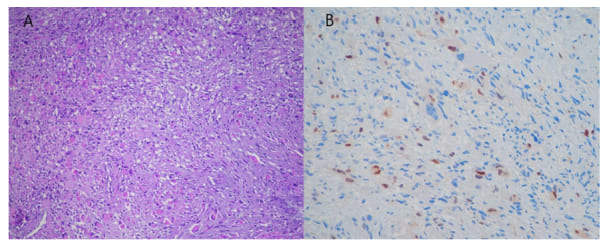
Figure 4: Histologic examination of bioptic samples of parapharyngeal mass showed a solid proliferation of spindle-shaped atypical cells with zonal evident rhabdomyoblastic differentiation (A) also revealed by nuclear immunhistochemical expression of myogenin (B). H&E (A), immunoperoxidase (B).
|
Sporadic MTT |
NF1-related MTT |
||
|
Median Age (years) |
40-50 |
28-36 |
|
|
Gender |
F>M |
M>F |
|
|
Sites |
Trunk |
Head and Neck |
|
|
Cytogenetic abnormalities |
Unknown |
17p deletion |
|
|
Local recurrence rate |
22-43% |
||
|
Metastatic rate |
20-50% |
||
|
Overall Survival at 5 years |
5-20% |
||
|
MTT= Malignant Triton Tumor; NF1= neurofibromatosis 1; F=female; M=male. |
|||
Table 1: Differences between sporadic MTT and NF1-related MTT.
|
Interventions |
Disease |
Phase clinical trial |
Treatments |
Endpoints |
NCT |
|
ASTX727 (Cedazuridine + Decitabine) |
MPNST with PRC2 Loss |
Phase II |
ASTX727 is a combination of two drugs (cedazuridine and decitabine) that have been designed to target cancer cells with a PCR2 mutation and to disrupt the cells’ ability to survive and grow |
CBR |
NCT04872543 |
|
Tazemetostat |
Recurrent/ refractory or metastatic MPNST |
Phase II |
Tazemostat orally twice a day up to 2 years or until disease progression or unacceptable toxicity |
ORR |
NCT04917042 |
|
Nivolumab plus Ipilimumab |
NF1 and newly diagnosed MPNST |
Phase I |
Neoadjuvant Nivolumab plus Ipilimumab prior to standard therapy |
Safety; Feasibility; MTD |
NCT04465643 |
|
Vaccine therapy |
Recurrent or unresectablr MPNST |
Phase I |
Intratumoral administration of a NISExpressing derivative manufactured from a genetically engineered strain of measles virus |
MTD; Safety; PFS |
NCT02700230 |
|
Abbreviation: MPNST= Malignant Peripheral Nerve Sheath Tumors; CBR=Clinical Benefit Rate; ORR= Overall Response Rate; NF1= Neurofibromatosis type 1; MTD= Maximal Tolerated Dose; PFS= Progression Free Survival; |
|||||
Table 2: Ongoing clinical trials (to February 2024).
Conclusion
Due to the complexity and the rarity of the disease, the management of MTT should be performed in referral centers and every step of the process requires a multidisciplinary approach. Further clinical studies are needed to improve the diagnosis and treatment of this rare disease.
Acknowledgments: This study was partially supported by Ricerca Corrente grants no. 08067617 and 08067611, Fondazione IRCCS Policlinico San Matteo.
Disclaimer: The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- Basnet A, Sebastian JA, & Capo G. (2015). Malignant Triton Tumors in Sisters with Clinical Neurofibromatosis Type 1. Case Reports in Oncological Medicine, 2015: 1–6.
- Mae K, Kato Y, Usui K, Abe N, & Tsuboi R. (2013). A Case of Malignant Peripheral Nerve Sheath Tumor with Rhabdomyoblastic Differentiation: Malignant Triton Tumor. Case Reports in Dermatology, 5: 373–378.
- Kamran S. C, Howar S. A, Shinagare AB, Krajewski KM, Jagannathan J. P, et al (2013). Malignant peripheral nerve sheath tumors: Prognostic impact of rhabdomyoblastic differentiation (malignant triton tumors), neurofibromatosis 1 status and location. European Journal of Surgical Oncology, 39:46–52.
- Li G, Liu C, Liu Y, Xu F, Su Z, et al (2015). Analysis of clinical features and prognosis of malignant triton tumor: A report of two cases and literature review. Oncology Letters, 10: 3551–3556.
- Stasik C. J, & Tawfik, O. (2006). Malignant peripheral nerve sheath tumor with rhabdomyosarcomatous differentiation (Malignant triton tumor). Archives of Pathology & Laboratory Medicine, 130: 1878– 1881.
- Bruzzone E, Melloni I, Barra S, Orcioni G. F, & Cocito L. (2018). A rare case of intracranial malignant triton tumor arising in the middle cranial fossa: a case report and review of the literature. Folia Neuropathologica, 56: 229–234.
- Shetty P. K, Baliga S. V, & Balaiah K. (2012). Malignant triton tumor: a rare case. Indian Journal of Surgery, 75: 362–365.
- Jaing T, Chuang C. K, Jung S, Wu C, Tseng C, et al (2015). Malignant triton tumor of the cervical spine: Report of one case and review of the literature. Pediatrics and Neonatology, 56: 58–61.
- Velagaleti, G. V, Miettinen, M, & Gatalica, Z. (2004). Malignant peripheral nerve sheath tumor with rhabdomyoblastic differentiation (malignant triton tumor) with balanced t(7;9)(q11.2;p24) and unbalanced translocation der(16)t(1;16)(q23;q13). Cancer Genetics and Cytogenetics, 149: 23–27.
- Koutsimpelas, D, Brieger, J, Heinrich, U, Torzewski, M, Sommer, C, et al (2011). Cytogenetic analysis of a malignant triton tumour by comparative genomic hybridization (CGH) and review of the literature. European Archives of Oto-Rhino-Laryngology, 268:1391–1396.
- Woodruff JM, Chernik NL, Smith MC, Millett WB, Foote FW (1973) Peripheral nerve tumors with rhabdomyosarcomatous differentiation (malignant ‘Triton’ tumors) Cancer. 32:426–439.
- Barh A, Mukherjee B, Koka K, & Krishnakumar, S. (2019). Triton tumor of the orbit. Orbit, 39:418–421.
- Kim, S. T, Kim, C. W, Han, G. C, Park, C, Jang, I. H, Eog, H, Choi, G, & Lee, H. (2001). Malignant triton tumor of the nasal cavity. Head & Neck, 23: 1075–1078.
- Zhou, B, Zhan, C, Tian, Y, Gao, Z, & Yan, S. (2024). Primary hepatic malignant triton tumor mimicking hepatocellular carcinoma by demonstrating arterial-phase hypervascularity and subsequent washout on dynamic contrast-enhanced imaging: a case report and literature review. Frontiers in Medicine, 11.
- Ram R, Gardne J. M, Alapati S, Jambhekar K, Pandey T, et al (2017). Malignant triton tumor (Malignant peripheral nerve sheath tumor with rhabdomyoblastic differentiation) occurring in a vascularized free flap reconstruction graft. International Journal of Surgical Pathology, 25: 462–467.
- Li Y, Peng Q, Jiang N, Molloy D. P, Zeng C, et al (2022). Computed tomography imaging features of malignant ‘triton’ tumor to facilitate its clinical diagnosis: report of two cases. BMC Medical Imaging, 22.
- Pemov A, Hua Li, William P, Margaret R. W, and David T. M, (2019) ‘Genetics of Human Malignant Peripheral Nerve Sheath Tumors’, Neuro-Oncology Advances,2019: I50–61.
- Bian Y, Xiang Y, Zhou X, Zhao D, Wu H, et al .(2019). A series of 10 malignant triton tumors in one institution. Medicine, 98: e16797.
- Grobmyer, S. R, Reith, J. D, Shahlaee, A. H, Bush, C. H, & Hochwald, S. N. (2008). Malignant peripheral nerve sheath tumor: Molecular pathogenesis and current management considerations. Journal of Surgical Oncology, 97: 340–349.
- Wong W. W, Hirose T, Scheithauer B. W, Schild S. E, & Gunderson L. L. (1998). Malignant peripheral nerve sheath tumor: analysis of treatment outcome. International Journal of Radiation Oncology Biology Physics, 42: 351–360.
- Enzinger FM, Weiss SW. (1988) Malignant tumors of peripheral nerves. In: Enzinger FM, Weiss SW, editors. Soft tissue tumors. St. Louis: C.V. Mosby; 1988: 781–815.
- Ducatman BS, Scheithauer BW, Piepgras DG, et al. (1986) Malignant peripheral nerve sheath tumors: A clinicopathologic study of 120 cases. Cancer 57:2006–2021.
- Raney B, Schnaufer L, Ziegler M (1987) Treatment of children with neurogenic sarcoma. Cancer 1987; 59:1–5.
- Moretti, V. M, Crawford, E. A, Staddon, A. P, Lackman, R. D, & Ogilvie, C. M. (2011). Early outcomes for malignant peripheral nerve sheath tumor treated with chemotherapy. American Journal of Clinical Oncology, 34: 417–421.
- McConnell Y. J, & Giacomantonio, C. A. (2012). Malignant triton tumors complete surgical resection and adjuvant radiotherapy associated with improved survival. Journal of Surgical Oncology, 106: 51–56.
- Chaudhry I, Algazal T, Cheema A, Faraj A. A, Malki N. A, et al (2019). Mediastinal malignant triton tumor: A rare case series and review of literature. International Journal of Surgery Case Reports, 62: 115– 1190.
- Clark, Matthew A (2005) “Soft-tissue sarcomas in adults.” The New England journal of medicine 353: 701-11.
- Kamperis E, Barbetakis N, Asteriou C, Kleontas A, and Christoforidou V, (2013) “Malignant triton tumor of the chest wall invading the lung. A case report and literature review,” Hippokratia, 17: 277–280.
- Terzic A, Bode B, Gratz KW, Stoeckli SJ. (2009) Prognostic factors for the malignant triton tumor of the head and neck. Head Neck. 31:679– 688.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.