In Silico Design of SARS-CoV-2 Peptides to Use as Potential Biomarkers for the Diagnosis of COVID-19 in Mexican Population
by Sharon Pérez Balbas1,2, Javier R. Ambrosio Hernández1, Rocio Tirado Mendoza1*, Lilian Hernández Mendoza1*
1Laboratorio de Biología de Citoesqueleto y Virología, Departamento de Microbiología y Parasitología, Facultad de Medicina, Universidad Nacional Autónoma de México. Edificio A Segundo Piso, Circuito Interior sin número. Universidad Nacional Autónoma de México. CP 04510, CdMx, México
2Laboratorio de Inmunofarmacología. Unidad Profesional Interdisciplinaria de Biotecnología (UPIBI), Instituto Politécnico Nacional, México
*Corresponding authors: Rocio Tirado Mendoza, Departamento de Microbiología y Parasitología, Facultad de Medicina, Universidad Nacional Autónoma de México. CP04510. CdMx, México.
Lilian Hernández Mendoza, Departamento de Microbiología y Parasitología, Facultad de Medicina, Universidad Nacional Autónoma de México. CP04510. CdMx, México.
Received Date: 29 August 2024
Accepted Date: 10 September 2024
Published Date: 13 September 2024
Citation: Pérez, B.S., Ambrosio H.J., Hernández M.L., Tirado R. (2024) In Silico Design of SARS-CoV-2 Peptides to Use as Potential Biomarkers for the Diagnosis of COVID-19 in Mexican Population. Infect Dis Diag Treat 8: 266. https://doi.org/10.29011/2577-1515.100266.
Abstract
The first case of COVD-19 in Mexico was reported in February 2020. Since then, different methods have been used in the world, including Mexico to detect SARS CoV-2. However, a significant number of asymptomatic cases, diagnostic tests with false positive results and different circulating variants, each with distinct patterns, have been observed, affecting different ethnicities. Considering the unique characteristics of each ethnicity, we propose in this study the design of peptides by in silico analysis of the viral proteins S, N, E, M and NSP12 of SARS CoV-2 in the context of the major histocompatibility complex class I and class II specific to the Mexican population. A bioinformatic analysis by the platforms: Immune Epitopes Data Base (IEDB) and Vaxygen was made to determine the peptides. Furthermore, GISAD platform was used to obtain the viral sequences of the most frequent variants of SARS CoV-2 circulating in our country during February 2020 to March 2021. A total of 7034 peptide sequences of SARS CoV2 viral proteins were analyzed. We identified 17 peptides from protein S (D614G), 3 peptides from protein E, 6 peptides from protein M, 1 peptide from protein N (R203K and G204R), and 13 peptides from NSP12 (P323L). All selected peptides demonstrated adequate immunogenicity and the ability to interact with the most frequent MHC alleles in our population, achieving a population coverage of 97.78% hence, they were potential biomarkers for diagnostic tests to SARS CoV-2 in our population.
Keywords: In silico peptides; SARS CoV-2; Mexican population; MHC alleles; Population coverage
Introduction
The first case of COVID-19 was reported in Wuhan, China, at the end of 2019 and the etiological agent was identified as a new respiratory virus classified as SARS CoV-2, classified as a member of the Coronoaviridae family [1,2]. This new viral respiratory infection spread rapidly to China and the rest of the world during the first trimester of 2020 [1-3]. Since the beginning of the pandemic until now, the effective transmission and dispersion of the virus have led to the emergence of viral variants; some of which are more transmissible, virulent or can potentially evade immunity induced by previous infections [4]. The emergence of variants with mutations that could be different and/or accumulative has played a relevant role in the course of the disease, resulting in millions of cases with different symptoms, from severe pneumonia that has led to hundreds of thousands of deaths worldwide, to asymptomatic cases [2,5].
Global health authorities have mainly focused on the viral variants which showed changes at the spike protein domains, these mutations could modify the binding to the ACE2 receptor. Changes in both binding and fusion domains can modify the clinical impact of the virus, its ability to colonize the respiratory tract and its transmissibility [6]. In order to categorize the different variants of SARS CoV-2, the WHO (https://data.who.int/dashboards/covid19/ variants), established three categories according to risk analysis: i) variants of interest (VOI), ii) variants of public health importance or variants of concern (VOC), and iii) variants of consequences. The pattern of circulation of the viral variants was an enormous task to get an efficient diagnostic to avoid false negatives results or undetected asymptomatic cases. The identification and characterization of the different variants was done by molecular biology tools and were sequenced by bioinformatics programs [7,8].
However, the need for rapid diagnosis has led to the development of rapid tests for the detection of SARS CoV-2 based on antibody detection. Nevertheless, it is important to consider the relevance of the CD4+ T cell response [9]. In humans, the RBD (receptor binding domain) is also highly immunogenic for T cells, comprising a cluster of peptides recognized by 94% of individuals [9-11]; and either the T cells could generate responses to peptides from other viral structural and non-structural proteins, although the structural proteins S, membrane (M) and nucleoprotein (N) are the most prominent targets of SARS-CoV-2-specific CD4+ T cells (9,10). Furthermore, serum levels of PD-L1 have a prognostic role in COVID-19 patients and associated to COVID-19 pathogenesis [12], Also, PD-L1 is responsible for T cell activation, proliferation, and cytotoxic secretion [13]. PD-1’s expression on the surface of monocytes and its activation by binding the PD-L1 ligand, and lead to inhibiting the activity of CD4+ T-cells by inducing the secretion of IL-10 [14], as the latter is a key regulator of immune homeostasis [15].
According to the previously described, we aimed to design in silico peptides for use in diagnostic tests with the objective to detect symptomatic as well as asymptomatic cases. For this purpose, we suggest a cluster of viral peptides presented in the context of MHC to be used as biomarkers of SARS CoV-2 infection. In our country, the first case of COVID-19 was reported on February 28th. Considering the most frequently reported variants in Mexico and the prevalent MHC alleles in the Mexican population, we analyzed 7034 peptide sequences of SARS CoV2 viral proteins. Predictions were made using IEDB resources (Proteasomal cleavage/TAP transport/MHC class I combined predictor and T cell class I MHC immunogenicity predictor) for different peptides. The immunogenicity of each peptide was calculated (Vaxijen v2.0 online) and population coverage was calculated for the Mexican population (Population Coverage). We identified 17 peptides from protein S (D614G), 3 peptides from protein E, 6 peptides from protein M, 1 peptide from protein N (R203K and G204R), and 13 peptides from NSP12 (P323L). They all demonstrated adequate immunogenicity, as well as the ability to interact with the most frequent MHC alleles in the Mexican population, achieving a coverage of 97.78%. The coverage for the global population was 94.09%.
Material and Methods
GISAID: Sequence Searching of SARS CoV-2 Variants Circulating in the Mexican Population
The in silico design of viral peptides is based on sequence data of SARS-CoV-2 and its variants deposited in the GISAID data base (https://www.gisaid.org/CoV2020/). GISAID classifies the new SARS-CoV-2 variants into clades (GISAID - hCov19 Variants). These clades correspond to eight high-level phylogenetic groups from an early split of S and L, followed by an evolution of L into V and G, and later of G into GH, GR, and GV, and, more recently GR into G RY (GISAID - hCov19 Variants). We consulted a total of 7034 sequences between February 28th, 2020, to May 13th, 2021, selecting and grouping those with the most frequent mutations. Finally, using the Nextstrain tool (Nextstrain / ncov / gisaid / global / 6m), which classifies the variants into major clades once a new variant reaches a global frequency of 20% at any given time, we constructed a phylogenetic tree with the 7034 sequences reported for our population.
Identification of the Widespread MHC alleles in the Mexican Population
The MHC alleles were selected using the platform of Allele frequencies (http://www.allelefrequencies.net/). The criteria resolution for the MHC alleles was at least four digits (e.g. A*02.01) [16,17].
Epitope Prediction for T Cells
The prediction of epitopes for CD8+ T cells , was done for different peptides using resources from the IEDB (http:/tools.iebd.org/main/ tcell/), such as the Proteasomal cleavage/TAP transport/MHC class I combine predictor and T cell class I MHC immunogenicity predictor. Afterwards, the immunogenicity of each peptide was calculated (Vaxijen v2.0 online). The threshold for strong binding peptides (IC50) was set at 50 nM to determine the binding and interaction potentials of the CD8+ T-cell epitope peptide and the class I alleles of the major histocompatibility complex (MHC). We selected ten alleles from the most frequent alleles found in the Mexican population: A*02:01, A*24:02, A*68:01, B*35:01, B*39:05, B*40:02, B*51:01, B*44:03, B*07:02, B*15:01. These alleles were used with the IEDB algorithm to predict the best peptides of the previously selected sequences of SARS CoV-2 variants; the selected peptides were nine aminoacids (a.a) in length.
The epitopes for T cells CD4+ were selected using the IEDB MHC-II Binding Predictions tool (http:/tools.iebd.org/mhcii). The selected peptides were fifteen a.a. long, and we applied the algorithm for the prediction of the mainly dominants epitopes for MHC II. Based on the most frequent alleles in the Mexican population, we selected 10 of them: DRB1*01:01, DRB1*04:04, DRB1*04:07, DRB1*07:01, DRB1*08:02, DRB1*11:01, DRB1*14:02, DRB1*14:06, DRB1*15:01, DRB1*16:02.
Immunogenicity
The epitopes predicted to be presented by MHC class I were analyzed using the Class I immunogenicity tool (http://tools.idb. org/immunogenicity) and the parameters were selected according to pre-established considerations in the platform.
The epitopes predicted to be presented by MHC class II were analyzed with the CD4+T Cell Immunogenicity Prediction tool (httpp://tools.iedb.org/CD4epscore) [18-21], and the parameters were selected according to pre-established considerations on the platform.
The peptides were ordered sequentially along the viral protein.
Antigenicity
The selected epitopes to be used as candidate biomarkers were analyzed using the Vaxijen v2.0 platform (http://www.ddgpharmfac.net/VaxiJen/VaxiJen.html). The cut off value was ≥ 0.4.
Population coverage
The population coverage for T cells epitopes was calculated using the IEDB Population Coverage Tool http:// tools.iedb.org/ population [18]. On this platform, we submitted the set of epitopes with the best characteristics of IC50, immunogenicity, and antigenicity, either in conjunction or alone, with the most frequent class I and class II alleles in our population.
Results
Variants of SARS CoV-2 Circulating in the Mexican population from February 28th, 2020, to May 13th 2021
We downloaded a total of 7034 sequences of different SARS CoV-2 variants circulating in our population. These sequences were open access in the GISAID platform, which allowed us to group the viral variants in clades (Figure 1). Using these data, we established a correlation between the clades and the number of cases (viral sequences) corresponding to each clade (Table 1). Furthermore, using the Nextstrain tool, we obtained the phylogenetic tree and identified the corresponding lineage using the Pango network (Figure 2). Our analysis of the sequences of circulating viral variants in our population showed that the most frequent were S_ D614G with 7,008 sequences or cases: N_ R203K with 5,083 sequences or cases, N_ G204R with 5,067 sequences and NSP12_P323L with 7005 sequences. It is important to mention that a variant could express more than one mutation [22,23]. This analysis allowed us to understand the relevance of the diversity of variants circulating among the population and identify the most frequent ones. The results were used to select variants for the insilico design of peptides proposed as biomarkers for diagnostic tests.
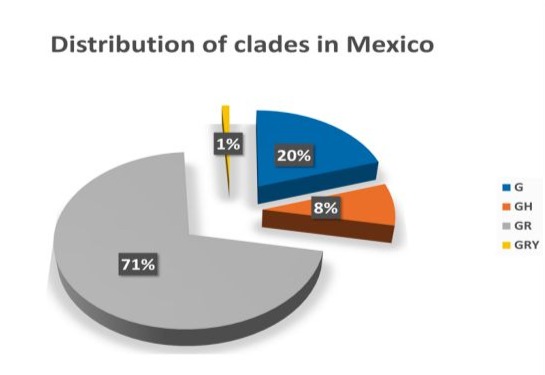
Figure 1: Distribution of the main clades in the Mexican population. A total of 7034 sequences from viral Mexican isolates were reported at the GISAID platform and all of them were classified in each clade. The clades with the highest circulation in our country were established based on these data.
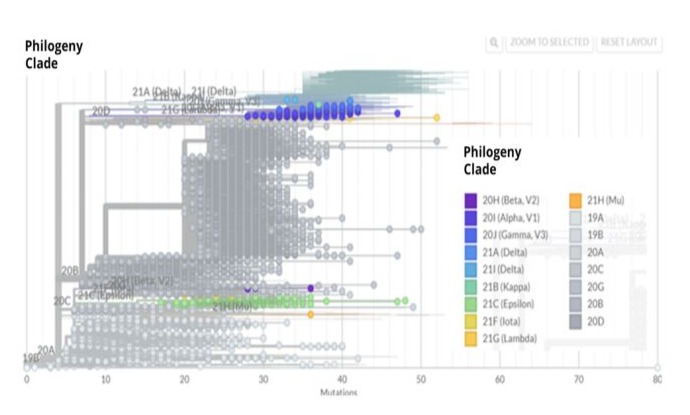
Figure 2: Phylogenetic tree of SARS CoV-2. The phylogenetic tree shows the 7034 sequences described up to May 13th, 2021. The construction of the phylogenetic tree was done with NextStrain (NextStrain (https: //nextstrain.org).
|
Clade |
Cases per mutation |
|
G |
1378 |
|
GH |
577 |
|
GR |
4939 |
|
GRY |
104 |
|
GV |
6 |
|
L |
2 |
|
S |
25 |
|
GK |
2 |
|
Total of cases |
7034 |
|
*Total number of cases from 28 February 2020 to 9 May 2021. |
|
Table 1: Distribution of Clades in Mexico.
The Most Frequent MHC alleles Found in the Mexican Population
The Mexican population possesses great genetic diversity. For the design of diagnostic tests, we considered the allelic frequencies of the most common HLA genes in the Mexican population, as HLA alleles are critical components in the viral antigen presentation pathway, and their ability to form the peptide-MHC complexes (pMHC). According to the Allele frequencies platform, our results show that the three most frequent class I alleles in our population were: A*02:01, A*24:02 and A*68:01 (Figure 3A). Among these molecules, the haplotype A*02:01 is particularly relevant as it has been reported as a risk factor to develop severe COVID-19 [24,25]. Meanwhile, the molecules HLA-A*11:01 and HLA-A*24:02, were more efficient in presenting viral peptides, thereby eliciting a better CD8+ T cell response against SARS CoV-2. We also identified that the class I HLA alleles locus B were B *35.01, B*39.01 and B*40:02 (Figure 3B). Some reports suggest a relative association between the B*35.01 allele locus and a low risk of developing severe COVID-19. This data is relevant for us because this allele is the most prevalent in our population.
The immune response mediated by CD8+ T cells is without a doubt fundamental in the resolution of viral infections, and COVD-19 it is no exception. Thus, we determined the distribution of class II HLA molecules in our population. The results indicated that the most frequent were DRB1*04 ≥, DRB1*08 ≥, DRB1*14 ≥, DRB1*07 ≥, DRB1*13 ≥, DRB1*11≥, DRB1*01≥, DRB1*15 ≥, DRB1* 03:01≥ and DRB1*16 (Figure 3C). Identifying the most common class I and II alleles allowed us to select candidate peptides in silico to be used as biomarkers for COVID-19 diagnosis.
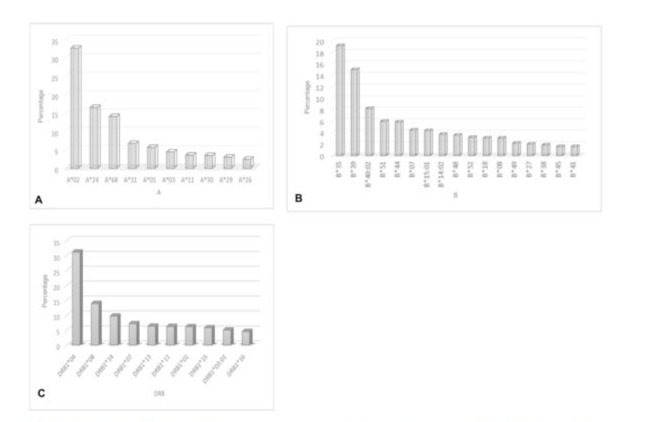
Figure 3: Distribution of HLA class I and II in the Mexican population. The most frequent HLA alleles in the Mexican population were obtained using the Allele frequencies platform (http://www.allelefrequencies.net/). The selection of the HLA molecules were on the basis of a resolution of at least 4 digits. A. Distribution of HLA class I locus A, the most frequent alleles were A*02, A*24, A*68. B. Distribution of HLA class I locus B molecules, the most frequent alleles were B*35, B*39, B*40:02. C. Distribution of class II HLA molecules, the most frequent alleles BRB1*04, DRB1*08, DRB1*14.
In Silico Design of SARS CoV-2 Peptides starting from Sequences of Mexican Viral isolates Reported in GISAID
For the design of the in silico peptides, we selected viral sequences from the Mexican isolates reported in the GISAID platform. From these sequences, the viral proteins expressing the most frequent mutations reported in our population were selected. Among them were the Spike protein (S D614G), Nucleoprotein (N R203K; NG204R), Envelope protein (E), Matrix protein (M) and the viral polymerase nonstructural protein (NSP-12 P323-L). We analyzed nearly 300000 peptides. These peptides were selected based on criteria from IEDB and Vaxijen, such as affinity (≤ 50nM), processing and presentation (≤ 1) n in the context of HLA class I. We obtained 13 peptides for the S D614G protein; 4 peptides for the E protein; 4 peptides for the M protein, One for N protein (N R203K; NG204R) and 17 for NSP12 P323L protein. Moreover, the selected peptides were validated using the Immunogenicity Tool (http://tools.iedb.org/immunogenicity), confirming that these peptides were the candidates with the best immunogenic and antigenic capacity.
Considering the parameters established by the platform (http:// tools.iedb.org/immunogenicity), we chose peptides in the context of HLA class I, our results showed in the context of HLA-B*35:01 two peptides (LPFNDGVYF and WPWYIWLGF) for the S protein (D614G), for the E protein, we chose one peptide (FLAFVVFLL) in the context of HLA-A*02:01, for the N protein, the sequence of the peptide was SPRWYFYYL in the framework of HLA-B*07:02.; the corresponding peptide for the M protein was LVIGAVVILR with the molecule HLA-A*68:01, and finally two peptides (TSFGPLVRK and VVSTGYHFR) for the NSP-12_ P323L, both peptides were presented by HLA-A*68:01 (Table 2).
We also designed the peptides that were presented by HLA class II. For the selection of these peptides, we used the platform http://tools. iedb.org/main/tcell/, establishing parameters of immunogenicity ≥ 50 and antigenicity ≤ 0.4. The results demonstrated that the best candidates were peptides corresponding to the M protein (FIASFRLFARTRSMW, IASFRLFARTRSMWS and ASFRLFARTRSMWSF), all of which bind to HLA-DRB1*11:01 (Table 3).
Finally, our peptides designed by IEDB and Vaxijen, were proposed as candidates to be used as markers for diagnostics in our population in the context of the most frequent HLA class I and Class II in the Mexican population (Table 2 and 3).
Population coverage
All the selected peptides met the parameters established by the different tools of the IEDB platform and Vaxijen. These peptides were selected to be used as biomarkers for diagnostic tests, in the context of HLA class I and Class II, with the objective of reducing the false-negative results. Moreover, these data allowed us to determine the population coverage, which estimates the percentage of individuals within a population (Mexico or worldwide) likely to have at least one in silico designed epitope expected to interact with their HLA class I and II alleles. Moreover, it was considered that the epitopes could be processed as the cells carried out this biological process. The set of peptides evaluated, associated with the most frequent HLA class I alleles in our population, provided an 86.41% population coverage. The same analysis indicated that this set of peptides could be recognized by the world population with a coverage percentage of 76.67%. (Figure 4). For HLA class II epitopes, it was observed that they cover 83.66% of the Mexican population and 76.69% of the world population (Figure 4). Finally, we analyzed the coverage of the epitopes of HLA class I and II, and the results demonstrated that both the Mexican population and the world population were covered at over 90% (Figure 4).
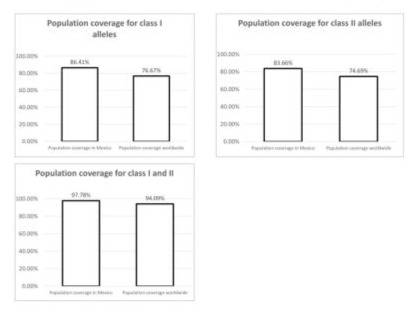
Figure 4: Population coverage of HLA class I epitopes. A. Population coverage indicates the percentage of the T-cell epitope set of the SARS-CoV S, N, M, E, NSP12_p323L interacting with the most frequent HLA class I alleles in the Mexican (86.41) and global (76.67%). Population coverage of HLA class II epitopes. T-cell epitope set S, N, M, E, NSP12_p323L for SARS-CoV-2 for Mexican (83.66%) and global (74.69%). C. Population coverage of HLA class I and class II epitopes. Set of epitopes of S, N, M, E, NSP12_p323L of SARS-CoV-2 for the Mexican (97.78%) and global (94.09%) population coverage worldwide (94.09%).
Discussion
SARS-CoV2 causes a life-threatening disease called COVID19, which can be particularly severe in individuals with comorbidities. The effective transmission and dispersion of the virus have led to the emergence of various viral variants. Some of these variants are more transmissible, virulent or can potentially evade immunity induced by vaccination or previous infection. In Mexico, according to GISAID data up to May 13, 2021, the most frequent clades were GR, G, GH and GRY. Using the NextStrain platform, the most prevalent lineages in our country were identified as B.1.1.519 and B.1, which correspond to the GR and GH clades, respectively.
In the case of the sequences reported in GISAID, there were a total of 4939 for the GR clade, 1378 for the G clade, 577 for the GH clade, and finally 104 for the GRY clade. All these sequences share the D614G mutation located in the receptor-binding domain (RBD), which is near the proteolytic cleavage of the S1 subunit. This modification favors proteolytic processing and in consequence, viral entry.
Moreover, according to the reported sequences of the Mexican isolates, the most frequent mutation was RdRp P323L as shown in Table 2 and 3. This modification promotes a more efficient viral replication and likely improves the catalysis of the transcription of viral genomic and subgenomic RNAs. Furthermore, we could identify two other mutations at the nucleocapsid gene at two different sites: R203K and G204R. These modifications are relevant at two levels of the viral replication. The nucleocapsid protein plays a fundamental role during virion assembly through its interactions with the viral genome and the membrane protein M and has an important role in enhancing the efficiency of subgenomic viral RNA transcription and viral replication.
Although we have described the main mutations reported in the Mexican population, other mutations were also present, such as M Q19E; A63T and E T9I. For our study, all the sequences previously described were relevant for designing the viral peptides proposed as candidates for biomarkers in the diagnosis of SARS CoV-2 in the Mexican population.
For this purpose, we identified the most frequent class I alleles expressed in the Mexican population. According to our data, the following were found: HLA-A*01:01, HLA-A*02:01, HLA-A*03:01,HLA*24:02, HLA-A*26:01, HLA- A*31:01, HLA-A*68:01, HLA- HLA- B*18:01, HLA-B*35:03, HLA-B*38:01, HLA- B*44:02, HLA-B*44:03 and HLA-B*51:0. Among these, one of the most significant and commonly found is HLA-A*02:01, because, is considered a risk factor for developing severe COVID19 due to its deficient capacity for processing antigen presentation [25,26].
Meanwhile, HLA*11:01 and HLA*24:02 molecules play an important role in antigen presentation [27], with better processing of viral antigens and, consequently, a better antiviral T cell response [28]. The predictions for class I epitopes of SARS-CoV-2 proteins were identified using in silico methods and validated by IEDB and Vaxijen. From this analysis we found a total of 38 potential peptides. These peptides showed the best affinity characteristics (less than 50 nM), indicating good binding and interaction of the peptide with CD8+ T cells and the MHC I allele, with adequate processing assessed by the proteasomal cleavage tool / TAP / MHC class I transport, resulting in a high or low proteosome score combined with a good protein processing, peptides with the lowest scores were discarded.
We also included the locus B alleles of class I, the most frequently found in our population: HLA-B*18:01, HLA-B*35:01, HLA-B*38:01, HLA-B*44:02, HLA-B*44:03, HLA-B*51:01. Notably, the B*35, B*39 and B*40:02 molecules are relevant because HLA-B*35:01 is associated with a lower risk of developing severe COVID-19, as it shows a high capacity to present SARS-CoV-2 antigens compared to other HLA class I locus B alleles according to the literature [29]. This allele is also the most frequent, present in 19% of the Mexican population.
We also identified the most prevalent class II alleles in the Mexican population. Our study showed that DRB1*04, DRB1*08 and DRB1*14 were the most widespread. It has been described that DRB*04 is associated with increased susceptibility to CoV19, and DRB1*08 and DRB1*1 are more likely to be associated with severe COVID-19 [30-34], collectively representing a 19% of prevalence in Mexico. Furthermore, soluble HLA-G considered for early detection of gestational diabetes [35], could be considered since the latter has been associated with the severe clinical syndrome of SARS-CoV-2 infection [36].
Given the fundamental role of CD4+ T cell activation in the induction of an immune response, we identified the alleles with the highest frequency, among them were DRB*04, associated with increased susceptibility to CoVID-19, and DRB*08. HLA DRB1 shows a higher probability of susceptibility to severe CoVID-19, due to the involvement of MHC II involved in antigen presentation to CD4+ helper T cells to facilitate the humoral immune response, in relation to SARS-CoV-2 infection, HLA-DR levels are mainly related to monocytes, if their expression is low it is associated with a deregulated immune response.
Using this data, we designed and selected peptides with the best scores according to the parameters of both IEDB and Vaxijen platforms. We chose three epitopes of the M viral protein: FIASFRLFARTRSMW, IASFRLFARTRSMWS, and ASFRLFARTRSMWSF (same central/nuclear sequence “FRLFARTRS”). These epitopes were selected because of their immunogenicity and binding characteristics to the most frequent MHC molecules in our population, making them strong candidates for biomarkers for the development of diagnostic tests. The in silico analysis suggests that these peptides are likely to be processed and presented naturally, potentially inducing the production of neutralizing antibodies that can be used in diagnostic serological tests.
Since the peptides selected have the highest value for processing and presentation in the context of the most widely distributed MHCs in our population, and they are considered markers for the design of diagnostic tests, it is crucial to ensure a minimal margin of false negatives. Thus, population coverage was determined. Our combined population coverage data (HLA class I and class II) corresponds to 97.78% for the Mexican population. For the same peptides, the worldwide population coverage was 94.09%. This indicates that peptides designed based on the Mexican sequences reported in GISAID can be recognized by other populations and can be used in other countries.
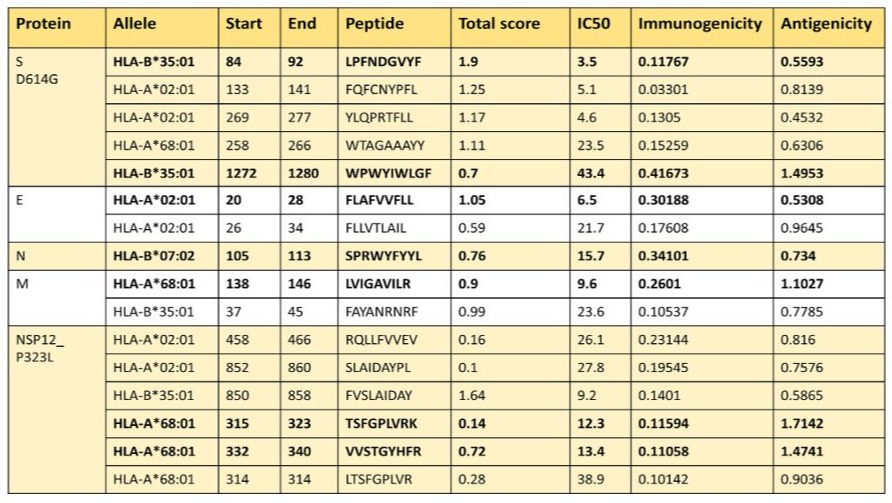
Table 2: Epitopes of the proteins S D614G, E, N, N, M NSP12_P323L for MHC I from the Mexican population.
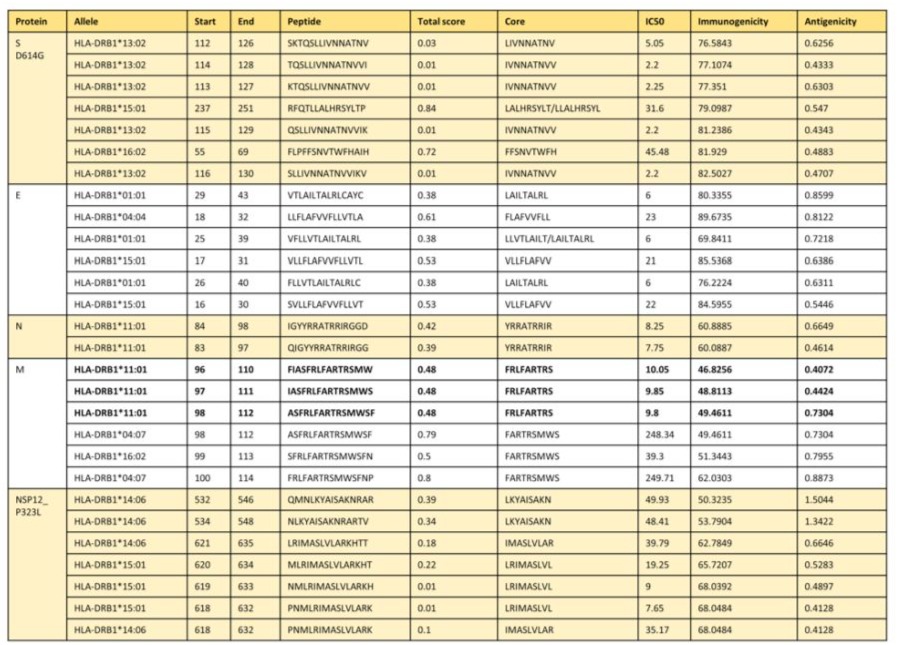
Table 3: Epitopes of the proteins S D614G, E, N, N, M NSP12_P323L for MHC II from the Mexican population.
Conclusion
In summary, a total of 7034 peptide sequences of SARS CoV2 viral proteins were analyzed and we could identify 17 peptides from protein S (D614G), 3 peptides from protein E, 6 peptides from protein M, 1 peptide from protein N (R203K and G204R) and 13 peptides from NSP12 (P323L). All of these peptides demonstrated adequate immunogenicity and the ability to interact with the most frequent HLA alleles expressed in the Mexican population. When the peptides for HLA I and HLA II molecules are tested together, the population coverage is 97.78% for the Mexican population, and the global population coverage is 94.09%. Due to their characteristics, we propose they would make strong candidates for biomarkers to be used in diagnostic tests.
Acknowledgments: The authors want to thank Dr. Diana Ríos for her assistance in the revision and preparation of the manuscript, ME Gisela Martinez, Q.F.B. Laura Chávez-Gómez, Q.F.B. César A. Rosales for their support to adjust the format of the figures, and Dr. Brenda Sandoval Meza for proofreading and editing the English version of the manuscript.
Funding: This research was funded by Dirección General de Asuntos del Personal Académico (DGAPA; IN-217519), UNAM, and Facultad de Medicina, UNAM.
Author contributions: Conceptualization, Rocio Tirado, Sharon Pérez Balbas and Lilian Hernández. Methodology, Sharon Pérez Balbas. Validation Sharon Pérez Balbas. Formal analysis, Rocio Tirado, Sharon Pérez Balbas and Lilian Hernández Mendoza.
Investigation, Sharon Pérez Balbas, Lilian Hernández and Rocio Tirado. Resources, Javier Ambrosio. Data curation, Rocio Tirado, Sharon Pérez Balbas and Lilian Hernández Mendoza. Statistical analysis, Sharon Pérez Balbas. Writing, Rocio Tirado. Supervision, Rocio Tirado and Javier Ambrosio. Project administration, Javier Ambrosio, and Rocio Tirado. Funding acquisition, Javier Ambrosio. All authors contributed to the article and approved the submitted version. Javier Ambrosio passed away by COVID-19 three years ago.
References
- Campi G, Perali A, Marcelli A, Bianconi A (2022) Sars-Cov2 world pandemic recurrent waves controlled by variants evolution and vaccination campaign. Sci Rep 12: 18108.
- Hu B, Guo H, Zhou P, Shi Z (2020) Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol 19: 141-154.
- Hui DS, Azhar EE, Madani TA, Ntoumi F, Kock R et al. (2020) The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health — The latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis 91: 264-266.
- Callaway, E (2021) Beyond Omicron: what’s next for COVID’s viral evolution. Nature 600: 204-207.
- Li R, Pei S, Chen B, Song Y, Zhang T et al. (2020) Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science 368: 489-493.
- Azkur AK, Akdis M, Azkur D, Sokolowska M, Van de Veen W et al. (2020) Immune response to SARS‐CoV‐2 and mechanisms of immunopathological changes in COVID‐19. Allergy 75: 1564-1581.
- Cantón R, De Lucas Ramos P, García-Botella A, García-Lledó A, Gómez-Pavón J et al. (2021) New variants of SARS-CoV-2. Revista Española de Quimioterapia 34: 419-428.
- Lauring AS, Hodcroft EB (2021) Genetic Variants of SARS-CoV-2— What Do They Mean? JAMA 325: 529.
- Torresi J, Edeling MA, Nolan T, Godfrey DI (2022) A Complementary Union of SARS-CoV2 Natural and Vaccine Induced Immune Responses. Front Immunol 13: 914167.
- Low JS, Vaqueirinho D, Mele F, Foglierini M, Jerak J et al. (2021) Clonal analysis of immunodominance and cross-reactivity of the CD4 T cell response to SARS-CoV-2. Science 372: 1336-1341.
- Sette A, Crotty S (2021) Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 184: 861-880.
- Sabbatino F, Conti V, Franci G, Sellitto C, Manzo V et al. (2021) PD-L1 Dysregulation in COVID-19 Patients. Front Immunol 12: 695242.
- Abdel Hafeez LA, Mansor SG, Zahran AM (2021) Expression of programmed death ligand-1(PDL-1) in Acute Myeloid Leukemia Patients and its relation to post induction Response. SECI Oncology Journal 9: 106-111.
- Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y et al. (2010) Programmed death-1–induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med 16: 452459.
- Mohammed D, Khallaf S, El-Naggar M, Abdel-Hameed MR, Bakry R et al. (2021) Interleukin-10: A Potential Prognostic Marker in Patients with Newly Diagnosed Multiple Myeloma. Research in Oncology. 17: 38-41.
- Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S et al. (2018) The Immune Epitope Data base (IEDB): 2018 update. Nucleic Acids Res 47: D339-D343.
- Tenzer S, Peters B, Bulik S, Schoor O, Lemmel C et al. (2005) Modeling the MHC class I pathway by combining predictions of proteasomal cleavage,TAP transport and MHC class I binding. Cellular And Molecular Life Sciences 62: 1025-1037.
- Bui H, Sidney J, Dinh K, Southwood S, Newman MJ et al. (2006) Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics 7: 153.
- Calis JJA, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD et al. (2013) Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput Biol 9: e1003266.
- Bui H, Sidney J, Peters B, Sathiamurthy M, Sinichi A et al. (2005) Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics 57: 304314.
- Can H, Köseoğlu AE, Alak SE, Güvendi M, Döşkaya M et al. (2020) In silico discovery of antigenic proteins and epitopes of SARS-CoV-2 for the development of a vaccine or a diagnostic approach for COVID-19. Sci Rep 10: 22387.
- https://iris.paho.org/bitstream/handle/10665.2/53217/ EpiUpdate11January2021_spa.pdf
- Pérez-Abeledo M, Sanz Moreno JC (2021) Variantes de SARS-CoV-2, una historia todavía inacabada. Vacunas 22: 173-179.
- Augusto DG, Hollenbach JA (2022) HLA variation and antigen presentation in COVID-19 and SARS-CoV-2 infection. Curr Opin Immunol 76: 102178.
- Trowsdale J, Knight JC (2013) Major Histocompatibility Complex Genomics and Human Disease. Annu Rev Genomics Hum Genet 14: 301-323.
- Weiner J, Suwalski P, Holtgrewe M, Rakitko A, Thibeault C et al. (2021) Increased risk of severe clinical course of COVID-19 in carriers of HLA-C*04:01. E Clinical Medicine 40: 101099.
- Khor S, Omae Y, Nishida N, Sugiyama M, Kinoshita N et al. (2021) HLA-A*11:01:01:01, HLA-C*12:02:02:01-HLA-B*52:01:02:02, Ageand Sex Are Associated with Severity of Japanese COVID-19 With Respiratory Failure. Front Immunol 12: 658570.
- Hensen L, Illing PT, Rowntree LC, Davies J, Miller A et al. (2022) T Cell Epitope Discovery in the Context of Distinct and Unique Indigenous HLA Profiles. Front Immunol 13: 812393.
- Jiang J, Natarajan K, Margulies DH (2019) MHC Molecules, T cell Receptors, Natural Killer Cell Receptors, and Viral ImmunoevasinsKey Elements of Adaptive and Innate Immunity. Adv Exp Med Biol1172: 21-62.
- Balas A, Moreno-Hidalgo MÁ, De la Calle-Prieto F,Vicario JL, Arsuaga M et al. (2023) Coronavirus-19 disease risk and protective factors associated with HLA/KIR polymorphisms in Ecuadorian patients residing in Madrid. Human Immunology 84: 571-577.
- Anzurez A, Naka I, Miki S, Nakayama‐Hosoya K, Isshiki M et al. (2021) Association of HLA‐DRB1*09:01 with severe COVID‐19. HLA 98: 37-42.
- Ebrahimi S, Ghasemi-Basir HR, Majzoobi MM, Rasouli-Saravani A, Hajilooi M et al. (2021) HLA-DRB1*04 may predict the severity of disease in a group of Iranian COVID-19 patients. Human Immunology 82: 719-725.
- Langton DJ, Bourke SC, Lie BA, Reiff G, Natu S et al. (2021) The influence of HLA genotype on the severity of COVID‐19 infection. HLA 98: 14-22.
- Schindler E, Dribus M, Duffy BF, Hock K, Farnsworth CW et al. (2021) HLA genetic polymorphism in patients with Coronavirus Disease 2019 in Midwestern United States. HLA 98: 370-379.
- Abdel Hameed MR, Ibrahiem OA, Ahmed EH, Sedky PR, Mohamed N et al. (2020) Soluble human leukocyte antigen-G evaluation in pregnant women with gestational diabetes mellitus. Egypt J Intern Med 32: 7.
- Apicella M, Campopiano MC, Mantuano M, Mazoni L, Coppelli A et al. (2020) COVID-19 in people with diabetes: understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol 8: 782-792.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.