Hyperuricemia and Gout as Uncommon Manifestation of Papillorenal Syndrome in an 18-Year-Old Young Man with PAX 2 Mutation: A Case Report and Review of the Literature
by Boutaleb Rajae*, Derwa Axel
Nephrology Department, Saint Jean clinic, Brussels, Belgium
*Corresponding author: Boutaleb Rajae, Nephrology Department, Saint Jean clinic, Brussels, Belgium
Received Date: 23 October 2024
Accepted Date: 28 October 2024
Published Date: 30 October 2024
Citation: Rajae B, Axel D (2024) Hyperuricemia and Gout as Uncommon Manifestation of Papillorenal Syndrome in an 18-Year-Old Young Man with PAX 2 Mutation: A Case Report and Review of the Literature. Ann Case Report. 9: 2035. https://doi.org/10.29011/2574-7754.102035
Abstract
Hyperuricemia is a metabolic abnormality, either acquired or inherited, characterized by elevated levels of uric acid (UA) in the serum. We are interested in the case report regarding hereditary hyperuricemia.
In this case, we are reporting on an 18-year-old man who experienced repeated gout attacks, hyperuricemia, and reduced fraction ejection of uric acid (FEUA). Additionally, he had renal anomalies such as proteinuria and renal hypoplasia and Focal and segmental glomerulosclerosis (FSGS) in the kidney biopsy and eye fundus revealed a bilateral optic disc pit .These findings prompted us to conduct a genetic study on the patient. To our surprise, the DNA sequence analysis did not detect mutations in UMOD or REN, which are commonly associated with familial juvenile hyperuricemic nephropathy (FJHN). Instead, a mutation in the paired box 2 (PAX2) gene was identified which is a very rare entity. We concluded that the patient had papillorenal syndrome (PAPRS).
Hyperuricemia is a rare and uncommon manifestation in patients with PAPRS and PAX2 mutation, and has only been documented in three studies in the literature. The diagnosis of PAPRS is often underestimated and not well-known by clinicians. It exhibits high clinical variability and phenotypic heterogeneity without well-defined diagnostic criteria. The presence of hyperuricemia should prompt consideration of this diagnosis and it should be included in the diagnostic criteria for PAPRS in the future.
Keywords: Hyperuricemia; Paired Box 2 (PAX2) Mutation; Optic Disc Pit; FSGS; Papillorenal Syndrome.
Abbreviation: UA : Uric acid; FEUA : Fractional extraction of uric acid; GFR: Glomerular Filtration Rate; UMOD: Uromodulin; PAX 2: Paired box 2; GWAS: Genome wide association studies; OMIM: Online Mendelin Inheritance in Man; ESRD: End stage of renal disease; MCKD: Medullary cystic kidney disease; FJHN: Familial juvenile hyperuricemic Nephropathy; CKD: Chronic kidney Disease; ENT: Ear nose throat; FSGS: Focal and segmental glomerulosclerosis; PAPRS: Papillorenal syndrome; VUR: Vesicoureteral reflux; CAKUT: Congenital kidney and urinary tractus; GLUT9: Glucose transporter; URAT1: urate transporter 1.
Introduction
Hyperuricemia is a metabolic abnormality, either acquired or inherited, characterized by elevated levels of uric acid (UA) in the serum. UA is primarily synthesized in the liver, intestines, and vascular endothelium as the product of purine metabolism. Purines can originate from the degradation of damaged, dying, and dead cells, either from exogenous or endogenous sources. The kidney plays a key role in excreting UA, eliminating approximately 70% of the daily produced UA through various molecules expressed in the renal proximal tubule, such as transporter 9 (GLUT9) and urate transporter 1 (URAT1). When UA production surpasses UA excretion, hyperuricemia occurs [1].
Acquired hyperuricemia may result from a purine-rich diet, certain medications, myeloproliferative disorders, obesity, and metabolic syndrome due to insulin resistance and its impact on reducing urinary urate levels [2,3]. Hereditary hyperuricemia can appear as an isolated non-syndromic disease or as part of a syndromic disorder. Genome-wide association studies (GWAS) have reported associations between hyperuricemia and around 30 loci. The Online Mendelian Inheritance in Man (OMIM) database lists approximately 20 syndromes associated with hyperuricemia, including Lesch–Nyhan syndrome, phosphoribosylpyrophosphate synthetase super activity, autosomal dominant tubulointerstitial kidney disease (previously known as medullary cystic kidney disease , and familial juvenile hyperuricemic nephropathy (FJHN) ) [4,5]. FJHN is the most common genetic disorder associated to hyperuricemia, with a UMOD mutation caused in 25-85% of cases by a UMOD mutation, and less than 5% by a REN mutation [6,7].
Studies have shown that uric acid levels increase progressively with age typically starting at around 40 years old [8]. Hyperuricemia, and especially gout attacks in a young patient, are uncommon and may indicate a genetic cause.
We present a case of a young man with hyperuricemia, gout, reduced FEUA, and CKD. DNA sequence analysis did not detect mutations in UMOD or REN, but did find a mutation in the paired box 2 (PAX2) gene, which is extremely rare.
Case Presentation
We report the clinical case of an 18-year-old young man of Turkish origin, with no parental consanguinity, who presented to an internal medicine consultation for recurrent gout attacks, with three attacks in one year.
On medical history, the patient complained of snoring and daytime fatigue, but no other specific complaints or symptoms, such as neurological manifestations, articular manifestations apart from at the time of the gout attacks, cutaneous or ophthalmological manifestations, the patient did not take anti-inflammatory drugs and did not consume drugs, alcohol or tobacco. The clinical examination revealed WHO grade 1 obesity, with a body mass index of 32.08 kg/m2. The rest of the clinical examination was normal, including normal blood pressure and absence of edema of the lower limbs. ENT and neurological examinations revealed no hearing loss or neurological abnormalities. In terms of family history, the patient's grandfather had chronic kidney disease, specifically immunotactoid glomerulonephritis, and there was no history of kidney disease in other family members.
An initial laboratory investigations revealed hyperuricemia at 14.5 mg/dl (Normal Value (NV) = 3.5 -7.2 mg/dl) with a reduced UAEF of 4.5% (NV = 7.5 ± 1.8%), elevated serum creatinine of 1.3 mg/dl [NV = 0.5-1.2 mg/dl], proteinuria of 1900 mg/g creatinine (NV <300 mg/g) (Table 1). Various biological tests were conducted to exclude other causes of hyperuricemia and proteinuria, as well as other pathological conditions. The results were as follows: viral serology (HIV, Hepatitis B and C) was negative, C3 and C4 complement levels were normal, anti-DNA, anti-PLA2r, ANCA antibodies were negative, and urine sediment was negative. There was no hypergammaglobulinemia or monoclonal peak on plasma protein electrophoresis, and no diabetes. Only a slight disturbance of liver enzymes was observed.
Imaging revealed hepatic steatosis and moderate hepatomegaly on abdominal ultrasound, the kidneys were small, the right kidney measuring 86x28 mm and the left kidney 84x26 mm (normal size is 95-110 x 32-38 mm). A PET-CT scan was performed to rule out malignant pathology as the cause of the hyperuricemia, but the result was negative.
Regarding the eye examination, the fundus showed bilateral inferotemporal excavation of the optic disc or optic disc pits, with a sharp papilla, a normal macula, and unremarkable blood vessels and periphery as depicted in Figures 1&2.
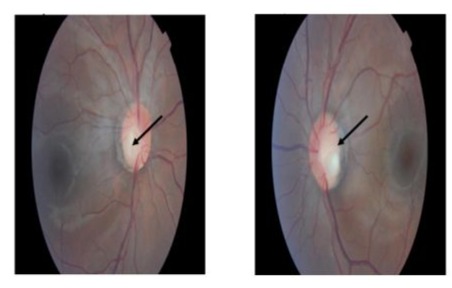
Figures 1&2: Eye fundus showing optic disc pit inferotemporal of the both eyes.
-A right renal biopsy was performed, yielding 10 glomeruli, a light microscopy revealed a segmental and focal glomerulosclerosis (FSGS) with no chronic lesions and no evidence of immune-mediated glomerulonephritis, morphological classification of FSGS (COLOMBIA): NOS (Figure 3).The electron microscopy showed podocyte foot process effacement typical lesion found inFSGS (Figure 4) and the immunofluorescence: IgA, IgG, IgM, C3, C1q was negative .
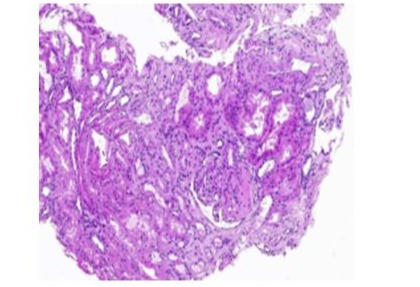
Figure 3: light microscopic study of kidney biopsy showing FSGS.
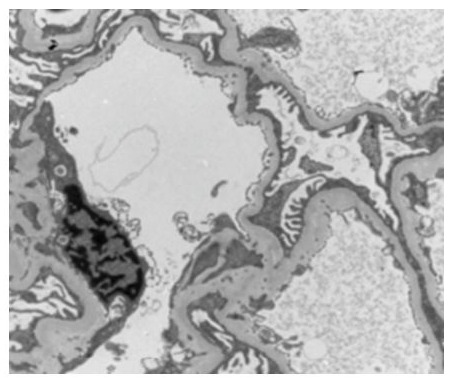
Figure 4: Electron microscopy study of kidney biopsy showed podocyte foot process effacement.
Given all the anomalies found in our patient, such as sub nephrotic proteinuria, symptomatic hyperuricemia with several gout attacks, renal hypoplasia, renal biopsy in favour of FSGS, and ophthalmic anomalies, a genetic study was conducted. The genetic sequencing confirmed the absence of UMOD and REN abnormalities. Additionally, no abnormalities were found within the SEC61A1 and HNF-1β genes, which have been reported to be associated with FJHN. Instead, a heterozygous nucleotide substitution was identified: c.310 C>T, leading to a missense mutation (p.Arg104*) in the PAX2 gene. This mutation involves the substitution of an arginine codon by a stop codon at position 104, thus interrupting the reading frame and leading to the production of a shortened protein. This mutation has already been described in the literature and is pathogenic.
|
Clinical and biological investigations |
Results |
Normal Range |
|
Serum creatinine ( mg/dl ) |
1,4 |
0,5-1,2 |
|
EstimatedGFR (CKDEPI)ml/min/1,73m |
70 |
>90 |
|
P/C Ratio ( mg/g ) |
1950 |
<300 |
|
Uric acid(mg/dl) |
14,5 |
3,5 -7,2 |
|
Gout |
+ |
|
|
Fe UA (%) |
4,5 |
7,5+/- 1 ,8 |
|
Renal ultrasonography (mm × mm) |
Right :86x28 Left : 84 X27 |
95-110 x32-38 |
|
Eye fundus |
Optic disc pits bilateral |
|
|
Gene study |
heterozygous mutation in PAX2c.310 C>T |
|
|
Kidney biopsy |
FSGS |
|
|
Note: + = present ; Abbreviation: GFR : glomerular filtration rate ; CKD-EPI : chronic kidney disease epidemiology collaboration (CKD-EPI) formula ; P/C ratio : proteinuria to creatinuria ratio ; FEUA :fractional excretion of uric acid , PAX2 : Paired box 2; FSGS : Focal and segmental glomerulosclerosis |
||
Table 1: Relevant clinical and biological abnormalities in the patient.
The patient was treated with medication to correct the metabolic disorders caused by chronic kidney disease (CKD) and high levels of uric acid. The progress of the CKD was monitored closely, and the patient is currently stable with medical therapy. However, close follow-up is being maintained to ensure prompt intervention if there are any changes in the patient's clinical condition. The patient also underwent regular follow-up appointments for ophthalmology and hearing tests.
The treatment focused on reducing protein filtration and uric acid levels by using Renin-angiotensin system inhibitors and hypouricemic medication. Additionally, the patient was advised to reduce protein, salt, and purine intake in their diet. Weight loss and regular physical exercise were strongly recommended for the patient. Management of the disease was focused on preventing ESKD and vision loss. Strategies include blood pressure control, avoidance of nephrotoxic drugs, and prevention of retinal detachment.
Discussion
The PAX2 gene encodes a nuclear transcription factor located near bands q24 and q25 on chromosome 10. It is one of the nine PAX paired box genes that encodes a DNA binding protein. This protein is mainly expressed during early embryonic development of the kidney, eye, central nervous system, ear, urogenital tract, and pancreas. Current data shows that it plays a crucial role in embryonic kidney development by regulating differentiation and renal cell proliferation at various fetal stages [9,10].
The PAX2 gene mutations have been mostly reported in patients with Papillorenal syndrome (PAPRS) (OMIM 120330). This syndrome is an autosomal dominant disease characterized by congenital renal and optic nerve abnormalities associated with mutations of the PAX2 gene [11,12]. PAPRS was initially described in 1977 as renal coloboma syndrome and later renamed papillorenal syndrome (PAPRS) [13,14]. In a series of patients with PAPRS and proven PAX2 mutations, renal, eye abnormalities, and high-frequency hearing loss were found in 92%, 77%, and 7% of patients, respectively [11].
The mutations of the PAX2 gene in human and mouse models have been associated with various renal anomalies. The most commonly encountered malformation is renal hypoplasia, found in 65% of cases (in 114 of 173 affected individuals) as in our patient's case [11,15]. Kidney Hypoplasia is usually bilateral and characterized on ultrasound examination by hypoplasia (small size for age) and hyper echogenicity. In fact, the PAX2 mutation is the most common cause of renal hypoplasia because of the important role played by PAX 2 in renal organogenesis [16].Zhang et al. reviewed and summarized 46 cases of PAX2 mutation from 2000 to 2016to study the correlation between phenotype and genotype and all the cases had renal hypoplasia [17]. Nishimoto et al. suggested also that renal hypoplasia was part of the RCS that was caused by heterozygous mutations of the PAX2 gene [18].
Other renal findings include vesicoureteral reflux (VUR), renal cysts, and multicystic dysplastic kidneys, occurring in 15%, less than 10%, and 5% of patients, respectively. Renal failure is reported in approximately 15% of cases, while chronic kidney disease (CKD) stage 5 requiring a kidney transplant is common and has a range of onset from birth to greater than 75 years of age [11]. Most data are aggregated from individual case reports and small case series, the exact incidence of ESKD requiring kidney transplantation is not precisely known.
Recent discoveries have found that the PAX2 gene is re-expressed in nephropathy and acts as a suppressor of WT1, an important transcription factor of podocytes. Its mutation causes congenital nephrotic syndrome and FSGS [19]. In immune kidney diseases, Letavernier et al observed changes in podocyte phenotypes in focal segmental glomerulosclerosis (FSGS) and found that PAX2 is implicated in the pathogenesis of renal interstitial fibrosis and glomerulosclerosis [20]. Zhang et al found that in primary nephrotic syndrome, PAX2 expression in renal tubules in steroid-resistant children was significantly increased compared to steroid-sensitive children [21]. In 2014, PAX2 was found to be associated with adult-onset focal segmental glomerulosclerosis (FSGS) and was listed as a causative mutation of FSGS [22; 23]. In our case, the patient has proteinuria and FSGS in the kidney biopsy as shown in pictures 2-3. According to a recent Korean study, including 27 patients with PAX2 mutations detected from 2004–2022, 4 of them had FSGS [24].
Around 77% of patients with PAX2 mutations present optic nerve involvement. Affected individuals may have unilateral or bilateral optic disc anomalies ranging from an optic disc pit to a chorio-retinal coloboma. The most common alteration is optic nerve dysplasia, characterized by an enlarged optic disc with emerging peripheral vessels and cilioretinal vessels. Other described characteristics include a vacant optic nerve with a central excavation, absence of the central retinal artery, presence of multiple cilioretinal arteries in radial formation, retinal coloboma, and retro bulbar optic nerve cysts [11]. In our case report, the patient has bilateral optic disc pits, as shown in pictures 3 and 4. According to the study by Bower et al, this anomaly was found in less than 10% of patients. Other ophthalmoscopic findings include optic nerve coloboma, optic disc dysplasia, morning glory anomaly, and hypoplastic optic discs, occurring in approximately 50%, over 10%, 5%, and less than 5% of patients, respectively [11]. The Consequences of the ocular malformations include decreased visual acuity, blindness, and retinal detachment.
Zhang et al. reviewed and summarized reported cases of PAX2 mutation from 2000 to 2016, including 46 cases. They observed the reported phenotype and genotype of PAX2 mutation, as well as the different types of PAX2 mutations, such as frame-shift, missense, splice sites, and deletions found in patients with PAPRS. There are no clear genotype/phenotype correlations, and variable types of PAX2 located across 10 of the 12 PAX2 exons can lead to similar phenotypes. Additionally, the same mutation within members of the same family can have variable penetrance and manifestations of PAPRS [17]. This was confirmed by the Korean report, which performed a genotype-phenotype analysis in patients with PAX2 mutations [24]. Other less common abnormalities observed in patients with PAX2 mutations include neurosensory auditory loss (found in 7% of those affected), central nervous system malformations, cognitive delay, ligament laxity, and elevated pancreatic amylase levels [25].
Our patient presented with hyperuricemia, reduced FEUA, and recurrent gout attacks, which led to their consultation with the internal medicine department. This condition is rare and has been documented in patients with a PAX 2 mutation, with only three unrelated families reported in the literature. In 2013, Megaw et AL reported a case of a family with a PAX2 frameshift mutation, involving five affected males across three generations, all of whom experienced hyperuricemia and/or gout [26].
In 2019, Deng et al. reported a study involving 10 pediatric patients with a de novo heterozygous mutation of PAX2, resulting in a missense Arg140Trp mutation. This mutation was identified in a 14.8-year-old girl who presented with hyperuricemia, gout, renal disease, and ophthalmic abnormalities consistent with PAPRS [27,28]. In 2020, Stevenson et al. reported a case involving a family of 9 patients, 5 of whom had the PAX2 mutation. Among them, 2 had hyperuricemia and reduced FEUA with gout [29].
The direct relationship between the PAX2 mutation and hyperuricemia is not yet clear and has not yet been studied. However, it can probably be explained by the presence of renal anomalies and agenesis encountered with a PAX2 gene mutation who plays a crucial role in embryonic renal development. Uric acid is primarily excreted by the proximal tubule, specifically by membrane transporters such as GLUT9, and URAT1 [30,31]. Urinary excretion of uric acid is therefore influenced by these factors.
Conclusion
Hyperuricemia does not seem to be a common symptom in patients with PAX2 mutations. Based on the literature, renal and ophthalmological abnormalities are the most frequently reported. However, hyperuricemia, especially in young patients, those with chronic kidney disease with no known cause, and/or family histories of kidney diseases, should alert clinicians, especially ophthalmologists, internists, and nephrologists to consider the possibility of a PAX2 mutation diagnosis. It is important to note that the diagnosis of PAPRS is underestimated and poorly known by clinicians. In conclusion, the term PAPRS should be replaced with PAX2-related disorder due to the significant clinical variability and phenotypic heterogeneity, the lack of correlation between phenotype and genotype, and the absence of well-defined diagnostic criteria of PAPRS of which hyperuricemia should definitely be included in the future.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
Consent for publication: Written informed consent was obtained from the patient for publication of this Case Report and any accompanying images.
Funding Declaration: The authors report no funding declaration
Research data declarations: This manuscript does not report data generation or analysis
References
- Shuangshuang Z, Wang Y, Cheng J, Huangfu N, Zhao R, et al (2019) Hyperuricemia and Cardiovascular Disease Curr Pharm Des. 25:700-709.
- Hyon K Choi, Atkinson K, Karlson EW, Willet W, Curhan G (2004) Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med 350:1093-103.
- Yael S, Schlesinger N (2016) Beyond Joints: a Review of Ocular Abnormalities in Gout and Hyperuricemia Curr Rheumatol Rep 18:37.
- Yang QO, Kottgen A, Dehghan A, Smith AV, Glazer NL, et al (2010) Multiple genetic loci influence serumurate levels and their relationship with gout and cardiovascular disease risk factors. Circulation-Cardiovascular Genetics. 3:523–530.
- Kottgen A, Albrecht E, Vitart V, Krumsiek J, Hundertmark C, et al (2013) M Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nature Genetics. 45:145–154.
- Devuyst O, Olinger E, Weber S, Eckardt K, Kmoch S, et al (2017) Autosomal dominanttubulointerstitial kidney 11 disease. Nature Reviews Disease Primers. 5:60.
- Bleyer AJ, Kidd K, Zivna M, Kmoch S (2017) Autosomal dominant tubulointerstitial kidney disease. Advances in Chronic Kidney Disease. 24:86–93.
- Bickel C, Rupperecht HJ, Blankenberg S, Rippin G, Hafner G, et al (2002) Serum uric acid as an independent predictor of mortality in patients with angiographically proven coronary artery disease.Am J Cardiol. 89:12-7.
- Narlis M, Grote D, Gaitan Y, Boualia SK, Bouchard M (2007) PAX2 and Pax8 regulate branching morphogenesis and nephron differentiation in the developing kidney. J Am Soc Nephrol. 18:1121–1129.
- Rothenpieler UW, Dressler GR (1993) Pax-2 is required for mesenchyme-to-epithelium conversion during kidney development. Development. 119:711–720.
- Bower M, Salomon R, Allanson J, Antignac C, Benedicenti F, et al (2012) Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum Mutat. 33:457–66.
- Bower MA, Schimmenti LA, Eccles MR (2007) Renal coloboma syndrome In:, editors. GeneReviews® ; 1993-2017.
- Karcher H (1979) The morning glory syndrome. Klin Monbl Augenheilkd 1979;175:835–840.
- Rieger G (1997) On the clinical picture of Handmann’s anomaly of the optic nerve morning glory syndrome? Klin Monbl Augenheilkd 170:697–706.
- Porteous S, Torban E, Cho NP, Chua L, McNoe L, et al (2000) Primary renal hypoplasia in humans and mice with PAX2 mutations: evidence of increased apoptosis in fetal kidneys of PAX21Neu +/− mutant mice. Hum Mol Genet. 9:1–11.
- Woolf AS, Winyard PJ. (2000) Gene expression and cell turnover in human renal dysplasia. Histol Histopathol. 15:159–66
- Zhang L, Zhao L, Zhang Y, Sun BC, Ma Q (2018) New PAX2 heterozygous mutation in a child with chronic kidney disease: a case report and review of the literature MC Nephrology 19:245.
- Nishimoto K, Iijima K, Shirakawa T, Kitagawa K, Satomura K, et al. (2001) PAX2 gene mutation in a family with isolated renal hypoplasia. J Am Soc Nephrol. 12:1769–72.
- Fukuzawa R, Eccles M, Ikeda M, Hata J (2003) Embryonal hyperplasia of Bowman's capsular epithelium in patients with WT1 mutations. Pediatr Nephrol. 18:9–13.
- Letavernier E, Bruneval P, Mandet C, Duong JP, Huyen JP, et al (2007) High sirolimus levels may induce focal segmental glomerulosclerosis de novo. Clin J Am Soc Nephrol. 2:326–333.
- Zhang HQ, Yi ZW, He XJ, Dang XQ, He QN, et al (2005) PAX2 expression in children with steroid-resistant primary nephrotic syndrome. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 30:597–600
- Vivante A, Chacham OS, Shril S, Schreiber R, Mane SM, et al. (2019) Dominant PAX2 mutations may cause steroid-resistant nephrotic syndrome and FSGS in children. Pediatr Nephrol. 34: 1607–1613.
- Barua M, Stellacci E, Stella L, Weins A, Genovese G, et al (2014) Mutations in PAX2 associate with adult-onset FSGS. J Am Soc Nephrol. 25: 1942–1953.
- Ji Hyun Kim, Ahn YH, Jang Y, Park E, Kim SH, et al (2019) Genotype-phenotype analysis in patients with PAX2 mutations: beyond renal coloboma syndrome .Research square.
- Schimmenti LA (2011) Renal coloboma syndrome. Eur J Hum Genet 19:1207–1212.
- MegawRD, Lampe A, Dhillon B, Yoshida S, Wright AF (2020) Papillorenal syndrome in a family with unusual complications. British Journal of Ophthalmology, 97: 945–946.
- Deng, H, Zhang Y, Xiao H, Yao Y, Liu X, et al (2019) Diverse phenotypes in children with PAX2-related disorder.Molecular Genetics & Genomic Medicine 7:e701.
- Negrisolo S, Benetti E (2023) PAX2 and CAKUT Phenotypes: Report on Two New Variants and a Review of Mutations from the Leiden Open Variation Database. Int J Mol Sci. 24:4165.
- Stevenson M, Pagnamenta AT,Reichart S, Philpott C, Lines KE, et al (2020) Whole genome sequence analysis identifies aPAX2 mutation to establish a correct diagnosis for asyndromic form of hyperuricemia. Am J Med Genet Part A. 182A:2521–2528.
- Matsuo H , Takada M , Ichida K, Nakamura T, Nakayama A, et al (2009) Common defects of ABCG2, a high capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population.Sci Transl Med. 1: 5ra11.
- Caulfield M.J , Munroe P.B,O'Neill D, Witkowska K, Charcher FJ, et al (2008) SLC2A9 is a high-capacity urate transporter in humans.PLoS Med. 5: e197.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.