Fabry Disease in Elderly Patients with Unexplained Renal Failure and Left Ventricular Hypertrophy? Two Case Reports
by Yi-Zhen Lu1,2, Chih-Jen Wu1-4, Hong-Mou Shih1,3,5, Feng-Chi Kuo3,6, Cheng-Jui Lin1,2,7*
1Division of Nephrology, Department of Internal Medicine, Mackay Memorial Hospital, Taipei, Taiwan
2Department of Medicine, Mackay Medical College, Taipei, Taiwan
3Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, Taiwan
4Graduate Institute of Medical Sciences and Department of Pharmacology, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan
5Graduate Institute of Physiology, College of Medicine, National Taiwan University, Taipei, Taiwan
6Division of Nephrology, Department of Internal Medicine, Taitung Mackay Memorial Hospital, Taitung, Taiwan
7Mackay Junior College of Medicine, Nursing and Management, Taipei, Taiwan
*Corresponding author: Cheng-Jui Lin, Division of Nephrology, Department of Internal Medicine, Mackay Memorial Hospital, No. 92, Sec 2, Zhongshan N. Rd., Zhongshan Dist., Taipei City 104, Taipei, Taiwan.
Received Date: 26 October 2024
Accepted Date: 30 October 2024
Published Date: 04 November 2024
Citation: Lu YZ, Wu CJ, Shih HM, Kuo FC, Lin CJ (2024) Fabry Disease in Elderly Patients with Unexplained Renal Failure and Left Ventricular Hypertrophy? Two Case Reports. Ann Case Report. 9: 2043. https://doi.org/10.29011/2574-7754.102043
Abstract
Fabry disease is a rare inherited disease whose incidence is likely underestimated. The heterogeneous symptoms, atypical variants and multiple comorbidities of this disease make early diagnosis challenging. We presented two elderly patients who visited our clinic for impaired renal function and proteinuria which are easily regarded as complications of underlying diseases. Both had left ventricular hypertrophy and were ultimately diagnosed with later-onset Fabry disease. Both patients received enzyme replacement therapy thereafter and follow-up data showed improvements in disease severity and ventricular hypertrophy but still showed deteriorated renal function. Early diagnosis of Fabry disease may provide beneficial effects before irreversible changes occur in organs and improve patient quality of life. It is important to consider the diagnosis of Fabry disease when patients have renal failure and left ventricular hypertrophy, even in elderly patients with multiple comorbidities.
Keywords: Fabry Disease; Elderly Patients; Renal Failure; Proteinuria.
Introduction
Fabry disease (FD), also called Anderson-Fabry disease, is a rare inherited disease but is the second most prevalent lysosomal storage disorder after Gaucher disease [1]. The reported annual incidence is 1 in 100,000 which is likely underestimated [2]. The reported prevalence was estimated to be between 1:8,454 and 1:117,000 among males [3], and a study in our Center showed that the prevalence was approximately 0.59% in males with chronic kidney disease (CKD) [4].
FD is inherited in an X-linked manner caused by mutations in the GLA gene (Xq21.3-q22), resulting in deficient alpha-galactosidase A (a-Gal A) activity and leading to excessive deposition of glycosphingolipids in multiple organs including the eyes, skin, brain, heart, kidney, gastrointestinal tract and nervous system. Males are typically more severely affected than females are. Most females have a more variable disease course and may be asymptomatic. In addition, some atypical variants may present symptoms later in life. These heterogeneous symptoms of FD make it challenging to obtain a timely and correct diagnosis. The diagnosis of FD is based on the individual's clinical presentation and is typically confirmed by biochemical analysis (the serum level of alpha-galactosidase activity), molecular genetic testing and even biopsy. Here, we present two patients with atypical FD and their clinical courses:
Case Presentation
Patient 1
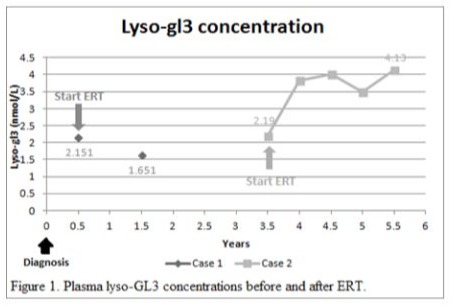
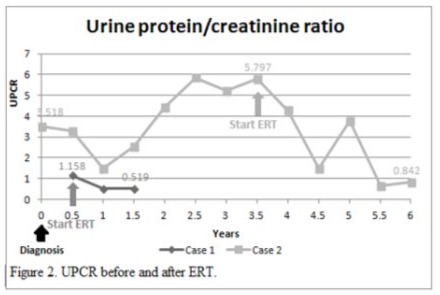
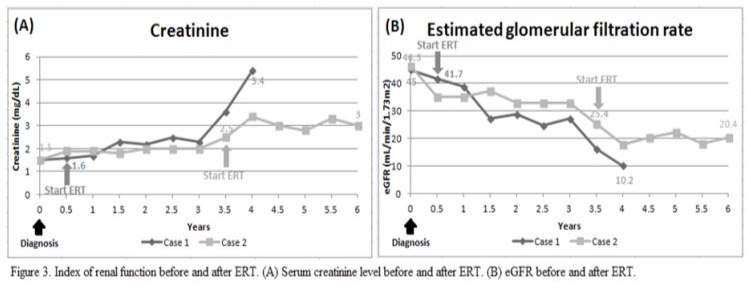
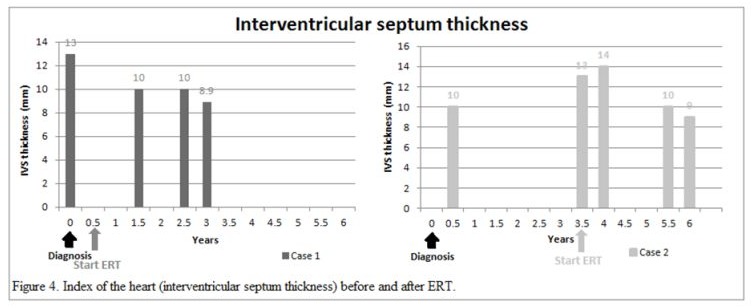
An 80-year-old man had underlying diseases of hypertension, hyperlipidemia, chronic ischemic heart disease, CKD stage 3 and gout. He received regular follow-up at the cardiologist’s clinic for nearly 10 years and was referred to the nephrology clinic for impaired renal function. His biochemistry showed serum blood nitrogen 18 mg/dl, creatinine (Cr) 1.5 mg/dl, urine protein 2+ and red blood cells 0/HPF. The daily protein loss was 1.472 g/day. Holter monitoring revealed episodes of idioventricular rhythm and premature ventricular contractions and episodes of nonsustained atrial fibrillation (Af) with slow ventricular response. A heart ultrasound showed concentric left ventricular hypertrophy (LVH), impaired diastolic function, and moderate mitral and aortic regurgitation. In addition, the thickness of the cardiac interventricular septum (IVS) was 13 mm (normal 7–11 mm). Cardiac magnetic resonance imaging (MRI) showed septal hypertrophy and intramyocardial fibrosis of the left ventricular wall. The FD screening test was positive. The genetic analysis revealed a mutation with the c.639+919 G>A mutation. He started to receive enzyme replacement therapy (ERT) after his final diagnosis. The plasma lyso-GL3 concentration, renal function and heart parameters were recorded before and after ERT (Figure 1-4). Family screenings were performed, and the patient’s daughter was diagnosed as a carrier, while her son was shown to have a cardiac variant type of FD. The patient was lost to follow-up after receiving ERT for approximately 3.5 years.
Patient 2
A 71-year-old man had underlying diseases of type 2 diabetes mellitus (DM), hyperlipidemia and angina for 17 years under medication control. Regular biochemical analysis revealed increased serum Cr (1.4 mg/dl) and decreased estimated Glomerular filtration rate (eGFR) (50 ml/min) beginning three years ago. His HbA1c level remained above 9% and he had severe albuminuria in recent years. He was referred to the nephrology clinic for persistent foamy urine and impaired renal function. Biochemical analysis revealed that the serum Cr concentration was 1.7 mg/dl and that the eGFR was 40 ml/min. Kidney echo revealed chronic renal parenchymal disease with a size of approximately 11.1×1.7 cm on the right and 13.3×1.5 cm on the left, consistent with diabetic nephropathy. The daily protein loss was 3.518 g/day. The FD screening test was positive. Further genetic analysis revealed a mutation, c.639+919 G>A. He suffered from a heart attack episode and was hospitalized at another hospital approximately one and a half years later. Cardiac catheterization revealed coronary artery disease, and two vessels were affected. He had received two stent placements. His EKG showed LVH according to the Cornell-voltage criterion. The heart echo showed grade I diastolic dysfunction and the IVS thickness was 10 mm. Cardiac MRI revealed intramyocardial fibrosis involving the lateral and inferoseptal segments of the basal left ventricle wall, consistent with cardiomyopathy related to FD. He underwent renal biopsy, which revealed diabetic nephropathy, Renal Pathology Society (RPS) class III, chronic tubulointerstitial nephritis and arterionephrosclerosis. Laminated, electron-dense lipid deposits were not found by electron microscopy. He also underwent endomyocardial biopsy, which revealed lamellar inclusions in the myocytes that were compatible with the features of FD. He started to receive ERT after the endomyocardial biopsy was performed. The plasma lyso-GL3 concentration, renal function and heart parameters were recorded before and after ERT (Figure 1-4). Family screening was suggested but was not performed due to personal reasons.
Discussion
Patients with FD may have variable presentations and disease courses according to their sex and specific GLA mutation. It has been more recently realized that later-onset phenotypic variants of FD have an approximately 5–10 times greater prevalence in the general population than the classic phenotype [5,6]. However, they are easily undiagnosed or have a delayed diagnosis due to the superimposed manifestation of various comorbidities, such as DM, CKD and coronary artery disease (CAD) in elderly people. Between our two patients, both had some common presentations and comorbidities. Patient 1 initially presented with ischemic heart disease along with concentric LVH found by heart echo and arrhythmia by Holter monitoring. Patient 2 had poorly controlled DM for a long time. Both patients presented to our clinic with progressive impairment of renal function and proteinuria, which are easily regarded as complications of underlying disease. However, genetic analysis of the GLA mutation revealed a missense mutation, c.639+919 G>A, in both patients, which confirmed the diagnosis of later-onset FD. A screening study of Taiwanese new-borns reported a dramatically greater incidence of GLA mutations (IVS4+919G) related to the cardiac variant of FD [7]. Early recognition of these patient groups in our country is prudent because earlier treatment with ERT may provide beneficial effects before irreversible changes occur in organs.
There are several clues that can help us to consider FD early, such as mild-to-moderate LVH in young adults and the presentation of CKD of uncertain etiology. For elderly patients with comorbidities, several specific characteristics suggest the possibility of FD. Cardiovascular manifestations of FD include LVH, aortic and mitral regurgitation, conduction defects, CAD, hypertension, and aortic root dilation. Notably, concentric ventricular hypertrophy is more common than asymmetric or eccentric hypertrophy, as shown in Patient 1. The LVEF is usually normal [8,9]. Right ventricular hypertrophy and impaired right ventricular function (as assessed by strain rate imaging) are also common [10]. Atrial arrhythmias are more common and conduction abnormalities may be caused by glycolipid deposition [11,12]. Patients with the cardiac variant may have mild to moderate proteinuria in the setting of normal or mildly impaired kidney function [13], as shown in Patients 1 and 2. Renal impairment often begins with microalbuminuria and proteinuria, which may be similar to the disease course of diabetic nephropathy. Initially, hyper filtration of the glomerulus could mask the impairment of renal function. A gradual decrease in renal function suggested damage to the glomerulus and renal tubules due to fibrosis, sclerosis and tubular atrophy. Eventually, the disease progresses to end-stage renal disease (ESRD). The significantly increased levels of urinary biomarkers of glomerular (especially type IV collagen) and tubular (especially N-acetyl-beta-glucosaminidase) dysfunction in adult patients with normal renal function and normoalbuminuria suggest that they may be biochemical indicators of an incipient stage in the natural history of kidney involvement in FD [14]. A specific histopathological pattern of FD can be identified by light microscopy, which includes glycosphingolipid accumulation in podocytes with numerous clear, uniform vacuoles in the cytoplasm causing a foamy appearance. On electron microscopy, deposits of Gb3 appear primarily within enlarged secondary lysosomes as lamellate membrane structures, called myeloid or zebra bodies; these deposits can be found not only in renal cells but also in cardiac myocytes.
In our study, both of patient received ERT with Fabrazyme® (agalsidase beta). Multiple indices including the plasma lyso-GL3 (Figure 1), urine protein and creatinine ratio (UPCR) (Figure 2), creatinine level and eGFR (Figure 3), and IVS thickness (Figure 4) were assessed before and after ERT. Previous studies in adults have shown that ERT reduces tissue deposition of Gb3 and may slow the decline in eGFR and reduce left ventricular wall thickness but has limited effect on patients with irreversible organ damage [15-17]. Long-term follow-up of our two patients after ERT treatment revealed that renal function indices, creatinine level and eGFR still deteriorated, while UPCR and Lyso-GL3 levels tended to decrease, suggesting improvement in disease severity. The potential cause of the failure of ERT to improve renal function could be related to advanced age and the fact that the patients presented to our outpatient clinic at a relatively advanced stage of CKD and other underlying diseases. The indices of the heart, IVS thickness, were measured by heart echo, which revealed a reduction in IVS thickness, which was consistent with the results of previous studies.
Conclusion
We present two cases of atypical FD with superimposed manifestations of multiple comorbidities. FD may be controlled and treated if diagnosed in time. In addition to performing detailed assessments of high-risk patients, such as patients with an unknown cause of LVH or family history of arrhythmia, conduction abnormality of the heart and CKD, peripheral neuropathy, ischemic stroke at younger age, we also need to be alert to patient with comorbidities but specific signs of atypical FD. Early diagnosis helps clinical physicians manage disease a timely manner via medical therapy and can improve patient prognosis and quality of life.
Declarations
Contributors: All authors contributed to planning, literature review and conduct of the review article. All authors have reviewed and agreed on the final manuscript.
Competing interests: None.
Patient consent for publication: Not applicable.
Ethics approval and consent to participate: Ethical approval for this publication by IRB at Mackay Memorial Hospital.
Availability of data and materials: Not applicable.
Funding: None from any funding agency in the public, commercial or not-for-profit sectors.
References
- Maria F, Peter J M, and John J H. (2006) Epidemiology of lysosomal storage diseases: an overview. Fabry Disease. Perspectives from 5 Years of FOS. Oxford PharmaGenesis.
- Dominique P G. (2010) Fabry disease. Orphanet J Rare Dis. 5: 30.
- Thaíza Passaglia B, Renato Demarchi F, Gianna Mastroianni K. (2020) Fabry disease: genetics, pathology, and treatment. Rev Assoc Med Bras, 66:s10-s16.
- Cheng-Jui L, Yin-Hsiu C, Thung-S L, Shih HM, Chen YC, et al. (2018) Results of Fabry Disease Screening in Male Pre-End Stage Renal Disease Patients with Unknown Etiology Found Through the Platform of a Chronic Kidney Disease Education Program in a Northern Taiwan Medical Center. Kidney Blood Press Res. 43:1636-1645.
- João Paulo O, Susana F. (2019) Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype– phenotype correlations. Appl Clin Genet. 12: 35–50.
- Tauben A, James A. W, and Nowell M. F. (2023) Anderson-Fabry disease cardiomyopathy: an update on epidemiology, diagnostic approach, management and monitoring strategies. Front Cardiovasc Med. 10: 1152568.
- Wuh-Liang H, Chien YH, Lee NC, Chiang SC, Dobrovolny R, et al. (2009) Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum Mutat. 30:1397-405.
- Kampmann C, Linhart A, Baehner F, Palecek T, Wiethoff CM, et al. (2008) Onset and progression of the Anderson-Fabry disease related cardiomyopathy. Int J Cardiol 130:367.
- Linhart A, Elliott PM. (2007) The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart 93:528.
- Niemann M, Breunig F, Beer M, Herrmann S, Strotmann J, et al. (2010) The right ventricle in Fabry disease: natural history and impact of enzyme replacement therapy. Heart 96:1915.
- Ikari Y, Kuwako K, Yamaguchi T. (1992) Fabry's disease with complete atrioventricular block: histological evidence of involvement of the conduction system. Br Heart J 68:323.
- Mehta J, Tuna N, Moller JH, Desnick RJ. (1997) Electrocardiographic and vectorcardiographic abnormalities in Fabry's disease. Am Heart J 93:699.
- Meehan SM, Junsanto T, Rydel JJ, Desnick RJ. (2004) Fabry disease: renal involvement limited to podocyte pathology and proteinuria in a septuagenarian cardiac variant. Pathologic and therapeutic implications. Am J Kidney Dis 43:164.
- Aguiar P, Azevedo O, Pinto R, marino J, Baker R, et al. (2017) New biomarkers defining a novel early stage of Fabry nephropathy: a diagnostic test study. Mol Genet Metab. 121:162–169.
- Ja Hye K, Beom Hee L, Cho JH, Kang E, Choi JH, et al. (2016) Long-term enzyme replacement therapy for Fabry disease: efficacy and unmet needs in cardiac and renal outcomes. J Hum Genet. 61:923-929.
- Germain DP, Elliott PM, Falissard B, Fomin VV, Hilz MJ, et al. (2019) The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol Genet Metab Rep. 19:100454.
- Weidemann F, Breunig F, Beer M, Sandstede J, Turschner O, et al. (2003) Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation. 108:1299-1301.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.