Endogenous Morphine, Dopamine, and GLP-1 Signaling: A Unified Evolutionary-Mitochondrial Framework for Reward, Addiction, and Neurodegeneration
by George B. Stefano¹*, Richard M. Kream²
¹Mind-Cell LLC, Baltimore, MD, USA
²ISL, Melville, NY, USA
*Corresponding Author: George B. Stefano, Mind-Cell LLC, Baltimore, MD, USA
Received Date: 16 April 2026
Accepted Date: 21 April 2026
Published Date: 23 April 2026
Citation: Stefano GB, Kream RM (2026) Endogenous Morphine, Dopamine, And GLP-1 Signaling: A Unified Evolutionary-Mitochondrial Framework For Reward, Addiction, And Neurodegeneration. J Surg 11: 11611 DOI: https://doi.org/10.29011/2575-9760.011611
Abstract
Endogenous morphine appears to represent a deeply conserved biochemical signaling system that predates full catecholamine diversification and remains functionally linked to dopamine-mediated reward, motivation, and motor activity. Because dopamine is both a central catecholamine transmitter and an essential intermediate in endogenous morphine biosynthesis, these signaling systems may be understood not as isolated processes but as components of a common evolutionary architecture. Emerging evidence indicates that Glucagon-Like Peptide-1 (GLP-1) signaling intersects with this architecture by modulating reward-related dopamine pathways while also stabilizing mitochondrial function and neuronal bioenergetics. Taken together, endogenous morphine, dopamine, nitric oxide, and GLP-1 signaling may form an integrated regulatory network that links energy acquisition, energy conservation, and adaptive behavior. Within this framework, addiction may reflect dysregulation of an ancient energy-reward axis, whereas Parkinson’s disease may reflect failure of a primordial dopamine-centered mitochondrial program affecting both motor and cognitive function. The persistence of these processes across phylogeny supports the view that complementary conformational stereospecificity and thermodynamically feasible molecular interactions have favored the retention of signaling solutions that enhance survival, behavioral efficiency, and cellular resilience.
Keywords: Cognition; Dopamine; Glp-1; Mitochondria; Morphine; Neurodegeneration; Parkinson’s Disease
Abbreviations: ATP: Adenosine Triphosphate; DA: Dopamine; ETC: Electron Transport Chain; GLP: Glucagon-Like Peptide-1; GLP-1R: Glucagon-Like Peptide-1 Receptor; NO: Nitric Oxide; ROS: Reactive Oxygen Species; μOR3 -Mu Opioid Receptor -Opiate Alkaloid Selective
Introduction
Endogenous morphine has moved from a provocative biological possibility to a recurrent theme in the literature on conserved cell signaling. Evidence for de novo morphine biosynthesis in animal cells, together with studies identifying dopamine as a key intermediate in that pathway, has supported the view that morphinergic processes are intrinsic to animal physiology rather than accidental reflections of dietary or plant exposure [1-5]. This proposition becomes more significant when considered alongside the hypothesis that the endogenous morphine synthetic pathway preceded, and later contributed to, catecholamine evolution, thereby linking morphine and dopamine within a shared biochemical ancestry [6,7]. In that sense, dopamine is not merely adjacent to endogenous morphine biology; it is built into its logic. This relationship suggests a broader interpretive shift. Dopamine has traditionally been treated as a catecholamine neurotransmitter governing movement, salience, reinforcement, and prediction, while morphine has been considered mainly in the context of analgesia, reward, and opioid pharmacology. Yet if dopamine is an endogenous morphine precursor and if morphine in turn modulates nitric-oxide-coupled mitochondrial processes, then reward, affective tone, metabolic restraint, and cellular energetics may be more unified than standard compartmentalized models suggest [8,7]. The present Opinion advances that integrative view and proposes that dopamine, endogenous morphine, nitric oxide, and GLP-1 signaling converge on a common evolutionary problem: how organisms acquire energy, regulate excitability, and preserve mitochondrial viability in fluctuating environments (Figure 1).
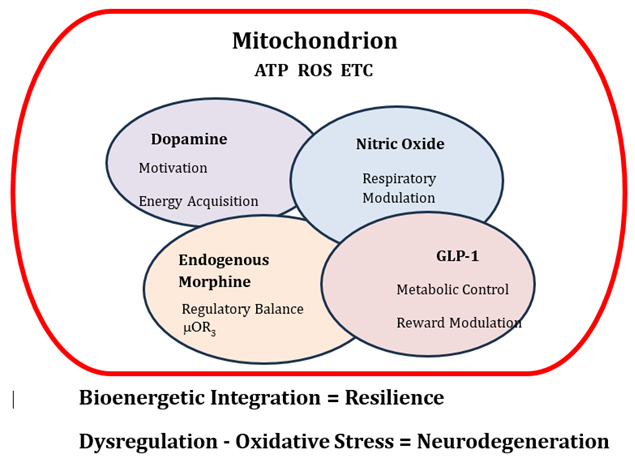
Figure 1: Integrated Dopamine–Morphine–GLP-1–Nitric Oxide Signaling Network Governing Mitochondrial Bioenergetics and Cellular Resilience. This conceptual schematic illustrates an integrated, dynamically weighted signaling network in which dopamine, endogenous morphine, Nitric Oxide (NO), and Glucagon-Like Peptide-1 (GLP-1) converge to regulate mitochondrial function and cellular energy homeostasis. The Venn-based architecture, with variable circle sizes, reflects the context-dependent and shifting influence of each signaling domain on mitochondrial activity. Dopamine is positioned as a primary driver linking motivational states and reward processing to energy acquisition demands. Endogenous morphine, biosynthetically derived from dopamine, functions as a homeostatic modulator that stabilizes neuronal excitability via μ-opioid receptor signaling, with downstream coupling to nitric oxide production. NO acts as a direct regulator of mitochondrial respiration by modulating electron transport chain activity, thereby fine-tuning oxygen utilization and constraining excessive Reactive Oxygen Species (ROS) generation. GLP-1 signaling is represented as a higher-order integrative axis that coordinates metabolic status with reward circuitry, attenuating excessive dopaminergic drive while enhancing mitochondrial efficiency, redox balance, and neuronal resilience. The mitochondrion is centrally positioned as the bioenergetic hub where these overlapping pathways converge to regulate ATP production and oxidative balance. The diagram emphasizes that adaptive flexibility within this network supports cellular resilience, whereas dysregulation—reflected by imbalance among these signaling systems—promotes mitochondrial dysfunction, increased oxidative stress, and heightened susceptibility to pathological states, including addiction and neurodegeneration.
An Evolutionary Logic of Energy Seeking and Energy Restraint
Dopamine’s role in mobility, motivation, and reward-directed behavior gives it an obvious adaptive value. An organism that can move, discriminate, anticipate, and persist in the pursuit of food gains a substantial survival advantage over one that cannot. In that sense, dopamine-linked motor and motivational systems can be understood as biological mechanisms for energy acquisition. They help convert environmental opportunities into usable metabolic substrates, including plant-derived carbohydrates such as glucose, and they thereby support both immediate survival and longer-term reproductive success [9]. This is especially important in ecological settings in which energy is unevenly distributed and successful acquisition requires not simply locomotion, but motivated locomotion guided by reinforcement. The evolutionary argument becomes stronger when one recognizes that random movement alone is metabolically expensive. Selection would be expected to favor signaling systems that transform undirected mobility into goal-oriented action, thereby reducing wasted expenditure while improving the probability of successful feeding and reproductive behavior. Dopamine fits that requirement. It energizes approach, incentive salience, and habit formation, but its deeper significance may be that it couples movement to a strategy of energy extraction from the environment. Because dopamine also participates in the morphine biosynthetic pathway, one may reasonably infer that motivational chemistry and morphinergic chemistry share a conserved ancestral basis rather than existing as wholly separate inventions [6,7].
Endogenous Morphine as A Modulator of Cellular Excitability and Mitochondrial Function
If dopamine helps organisms obtain energy, endogenous morphine appears well positioned to help organisms regulate how that energy is used. Morphine has been linked to highly selective mu-opiate receptor signaling, especially via truncated receptor variants associated with nitric oxide release, and this signaling has repeatedly been interpreted as down-regulatory, stabilizing, and protective in immune, vascular, neural, and progenitor cell systems [10,11]. In this framework, endogenous morphine does not simply suppress activity. Rather, it helps calibrate cellular responsiveness so that excitation remains productive rather than damaging [2]. This point becomes especially important at the mitochondrial level. Work on endogenous morphine and nitric oxide has suggested that morphinergic signaling can influence mitochondrial oxygen consumption, redox behavior, permeability transitions, and overall respiratory tone through nitric oxide-dependent processes [8,12-14]. Experimental evidence showing morphine-stimulated nitric oxide release from mammalian and human mitochondria supports a direct mechanistic relationship between morphinergic signaling and mitochondrial regulation [12]. Nitric oxide, in turn, is known to modulate cytochrome c oxidase activity and mitochondrial oxygen consumption in a reversible manner, providing a plausible route by which endogenous morphine could restrain excessive energy flux, limit reactive oxygen species generation, and preserve organellar integrity under stress [13-15]. Seen this way, endogenous morphine may serve as a biochemical counterweight to unconstrained excitation. Dopamine promotes the motivational and behavioral conditions necessary for energy acquisition; morphine, acting in concert with nitric oxide, helps ensure that energy expenditure and mitochondrial respiration do not escalate into damaging overactivity. This creates an elegant evolutionary complementarity between outwardly directed acquisition and inwardly directed stabilization. It also provides a rationale for why morphinergic processes would have been conserved even at very low concentrations: subtle regulation of excitability and mitochondrial output could offer disproportionately large survival benefits.
GLP-1 Signaling as A Convergent Reward-Bioenergetic Regulator
GLP-1 biology adds a modern and clinically relevant dimension to this conserved framework. Although GLP-1 is classically viewed as an incretin involved in glucose-dependent insulin regulation and feeding behavior, substantial evidence now indicates that GLP-1 receptors are expressed in mesolimbic structures such as the ventral tegmental area and nucleus accumbens, where they influence motivational and reward-related processing [16,17]. In those circuits, GLP-1 receptor activation reduces the rewarding value of palatable food and modulates dopaminergic tone in ways consistent with behavioral restraint rather than hedonic amplification [16,17]. That same logic appears to extend to drugs of abuse. Preclinical studies have shown that GLP-1 receptor agonism can attenuate alcohol-induced locomotor stimulation and accumbal dopamine release, reduce nicotine-associated reward behaviors, and suppress cocaine seeking or self-administration in rodent models [18-21]. More recently, a large cohort study in United States veterans with type 2 diabetes reported that GLP-1 receptor agonist use was associated with lower risks of several incident substance use disorders, strengthening the translational significance of the preclinical literature [22]. These observations are difficult to explain if GLP-1 signaling is regarded only as a peripheral satiety mechanism. They make better sense if GLP-1 is understood as part of a conserved system that dampens maladaptive reward intensity while improving metabolic efficiency. The mitochondrial dimension is equally important. GLP-1 receptor stimulation has been shown to preserve cortical and dopaminergic neurons in models of stroke and Parkinsonism, and broader reviews describe GLP-1 mimetics as neuroprotective agents that reduce oxidative stress, inflammation, and metabolic strain while supporting neuronal resilience [23,24]. Recent work has also emphasized mitochondria as a central locus through which semaglutide and related agents may influence neurodegenerative processes, appetite regulation, and addiction-relevant circuitry [25,26]. Thus GLP-1 signaling does not merely intersect dopamine at the level of synaptic reward. It also intersects the energetic substrate upon which dopaminergic neurons depend.
Toward A Unified Dopamine-Morphine-GLP-1 Hypothesis
The most parsimonious interpretation of these findings is that dopamine, endogenous morphine, and GLP-1 signaling participate in a partially overlapping evolutionary network that coordinates acquisition, valuation, and energetic stabilization. Dopamine drives approach, movement, reinforcement, and expectancy. Endogenous morphine tempers excitability and, through nitric oxide coupling, appears capable of directly modulating mitochondrial respiration and preserving metabolic homeostasis. GLP-1 signaling enters this scheme as a regulator that reduces maladaptive reward salience while improving mitochondrial and neuronal resilience. Together, these systems look less like separate biological stories and more like different layers of one conserved survival strategy. Within such a framework, addiction can be redefined as dysregulation of an ancient energy-reward axis. The addicted state is not simply excessive pleasure seeking. It is a condition in which dopaminergic drive becomes maladaptively amplified or distorted, morphinergic and nitric-oxide-dependent inhibitory calibration becomes insufficient or displaced, and mitochondrial efficiency progressively deteriorates under the burden of repeated metabolic and oxidative insult [27,28]. This helps explain why addictive processes are so persistent: they recruit systems originally optimized for survival and energy acquisition, then force them into a pathological loop. It also helps explain why GLP-1 receptor agonists may have therapeutic value in addiction. By reducing reward overvaluation and supporting mitochondrial integrity, they may restore balance to circuitry that has drifted away from its adaptive setting.
Conformational Complementarity, Mutation, and Thermodynamic Feasibility
The persistence of these pathways across phylogeny invites a more general evolutionary principle. Biological information is retained not only because genes are inherited, but because molecular interactions remain workable under thermodynamic constraints. Complementary conformational stereospecificity provides the practical grammar of this retention [29]. Ligands, receptors, enzymes, and signaling intermediates are preserved when their structural relationships continue to permit reliable and energetically feasible function. In this sense, beneficial mutations are not random improvements in abstraction; they are positive changes that remain compatible with an already workable biochemical architecture. The morphine-dopamine-GLP-1 nexus may therefore represent an example of evolutionary conservation grounded in structurally compatible solutions to the recurrent problem of energy management.
Parkinson’s Disease as Failure of A Primal Dopamine-Energy Program
The proposed framework also offers a distinctive perspective on Parkinson’s disease. Parkinsonism is often discussed in terms of dopamine deficiency, alpha-synuclein pathology, or selective neuronal vulnerability, and each of these perspectives remains important. Yet the substantia nigra dopaminergic neuron is also a metabolically stressed cell type whose autonomous activity, calcium handling, and large axonal arbor place unusual demands on linked purposeful movement, reinforcement, and cellular energetic support becomes progressively unstable. Mitochondria falter, oxidative burden rises, dopaminergic signaling wanes, and the organism loses both the drive and the capacity to execute adaptive behavior. This interpretation does not replace current Parkinson’s disease models, but it broadens them by locating dopamine loss within a more ancient survival architecture. It also helps explain why mitochondria-centered interventions, including GLP-1-based strategies, remain of interest in Parkinsonian disorders [23,24,26].mitochondrial performance [30,31]. If dopamine is a central organizer of movement and motivated energy acquisition, and if it is biochemically continuous with endogenous morphine pathways that influence mitochondrial regulation, then Parkinson’s disease may be interpreted as a breakdown in a primal dopamine-centered energy program rather than merely a transmitter deficit. Under this view, motor slowing, motivational blunting, and cognitive changes are different expressions of the same deeper imbalance. The system that once
Conclusion
Taken together, the available literature supports a coherent though still evolving hypothesis: endogenous morphine, dopamine, nitric oxide, and GLP-1 signaling form an interconnected regulatory system that couples reward, motivation, and mitochondrial energy metabolism. Dopamine provides the motivational engine by which organisms pursue and obtain energy. Endogenous morphine, through highly selective receptor processes and nitric oxide coupling, appears to moderate cellular excitability and influence mitochondrial respiration. GLP-1 signaling converges on both reward circuitry and mitochondrial resilience, reducing maladaptive reinforcement while stabilizing bioenergetic function. The shared logic of these systems is evolutionary economy. They help organisms acquire energy, avoid destructive overexcitation, and preserve the metabolic machinery required for survival. In this integrated model, addiction reflects the distortion of an originally adaptive energy-acquisition program, whereas Parkinson’s disease reflects collapse of a primal dopamine-linked mitochondrial strategy affecting both motor and cognitive domains. The broader implication is that reward biology cannot be fully understood without bioenergetics, and bioenergetics cannot be fully understood without the signaling systems that evolved to direct behavior toward energy acquisition while restraining cellular damage. Much remains to be clarified experimentally, but the hypothesis offers a biologically plausible and clinically suggestive framework for linking endogenous morphine, dopamine, GLP-1 therapeutics, and mitochondrial regulation in one evolutionary narrative.
References
- Gintzler AR, Levy A, Spector S (1976) Antibodies as a means of isolating and characterizing biologically active substances: presence of a non-peptide, morphine-like compound in the central nervous system. Proc Natl Acad Sci U S A 73: 2132-2136.
- Stefano GB, Scharrer B (1994) Endogenous morphine and related opiates, a new class of chemical messengers. Adv Neuroimmunol 4: 57-67.
- Poeaknapo C, Schmidt J, Brandsch M, Dräger B, Zenk MH (2004) Endogenous formation of morphine in human cells.Proc Natl Acad Sci U S A 101: 14091-14096.
- Kream RM, Stefano GB (2006) De novo biosynthesis of morphine in animal cells: an evidence-based model. Med Sci Monit 12: RA207-RA219.
- Zhu W (2005) Tyrosine and tyramine increase endogenous ganglionic morphine and dopamine levels in vitro and in vivo. Med Sci Monit 11: BR397-BR404.
- Stefano GB, Kream RM (2007) Endogenous morphine synthetic pathway preceded and gave rise to catecholamine synthesis in evolution. Int J Mol Med 20: 837-841.
- Stefano GB, Kream RM (2010) Dopamine, morphine, and nitric oxide: an evolutionary signaling triad. CNS Neurosci Ther 16: e124-e137.
- Kream RM, Stefano GB (2009) Endogenous morphine and nitric oxide coupled regulation of mitochondrial processes. Med Sci Monit 15: RA263-RA268.
- Stefano GB, Kream RM, Esch T (2023) Mobility Coupled with Motivation Promotes Survival: The Evolution of Cognition as an Adaptive Strategy. Biology 12: 80.
- Cadet P, Mantione KJ, Stefano GB (2003) Molecular identification and functional expression of mu3, a novel alternatively spliced variant. J Immunol 170: 5118-5123.
- Cadet P (2007) A functionally coupled mu3-like opiate receptor/nitric oxide regulatory pathway. J Immunol 179: 5839-5844.
- Stefano GB (2015) Morphine stimulates nitric oxide release in mammalian/human mitochondria. J Bioenerg Biomembr 47: 409-417.
- Giulivi C, Kato K, Cooper CE (2006) Nitric oxide regulation of mitochondrial oxygen consumption I: cellular physiology. Am J Physiol Cell Physiol 291: C1225-C1231.
- Kanai AJ (2001) Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria. Proc Natl Acad Sci U S A 98: 14126-14131.
- Brown GC, Cooper CE (1994) Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration.FEBS Lett 356: 295-298.
- Alhadeff AL, Rupprecht LE, Hayes MR (2012) GLP-1 neurons project to reward centers to control food intake.Endocrinology 153: 647-658.
- Dickson SL (2012) Exendin-4 decreases the rewarding value of food. J Neurosci 32: 4812-4820.
- Egecioglu E, Engel JA, Jerlhag E (2013) Exendin-4 attenuates the rewarding properties of alcohol. Addict Biol 18: 487-495.
- Egecioglu E (2013) GLP-1 analogue attenuates nicotine-induced reward behaviors. PLoS One 8: e77284.
- Hernandez NS (2019) GLP-1 receptor activation attenuates cocaine seeking. Neuropsychopharmacology 44: 431-439.
- Sorensen G (2015) GLP-1 receptor agonist reduces cocaine self-administration. Physiol Behav 149: 262-268.
- Cai M (2024) GLP-1 receptor agonists and risk of substance use disorders among US veterans. BMJ 386: e086886.
- Li Y (2009) GLP-1 receptor stimulation preserves neurons in stroke and Parkinsonism models. Proc Natl Acad Sci U S A 106: 1285-1290.
- Holscher C (2014) Central effects of GLP-1: opportunities for neurodegenerative disease treatment. J Endocrinol 221: T31-T41.
- Stefano GB (2025) The Anatomical and Evolutionary Impact of Pain, Pleasure, Motivation, and Cognition. Int J Mol Sci 26: 5491.
- Stefano GB (2026) Convergent Mitochondrial and Reward-Circuit Mechanisms Underlying Appetite, Addiction, and GLP-1 Therapeutics. J Surg 11: 11545.
- Stefano GB (2007) Nicotine, alcohol and cocaine coupling via endogenous morphine signaling. Med Sci Monit 13: RA91-RA102.
- Kream RM, Stefano GB (2010) Interactive effects of endogenous morphine, nitric oxide, and ethanol on mitochondrial processes. Arch Med Sci 6: 658-662.
- Stefano GB (1986) Conformational matching: a possible evolutionary force in signal systems. CRC Handbook. 1986.
- Surmeier DJ (2007) Calcium, ageing, and neuronal vulnerability in Parkinson’s disease. Lancet Neurol 6: 933-938.
- Surmeier DJ (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18: 101-113.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.