Development and Initial Design of the Hemagglutinin Stem Region in a Universal Influenza Vaccine
by Nikita Kuldyushev1, Irina Tcymbarevich2*, Marina Stukova3, Ekaterina Romanovskaya-Romanko3, Maria Sukhova2, Igor Krasilnikov2
1Research Center for Translational Medicine, Sirius University of Science and Technology, Olimpiyskiy ave. b.1, Sirius, Krasnodar region, 354340, Russia
2CJSC Biotechnology Developments, 119136 Moscow, Russia
3Smorodintsev Research Institute of Influenza of the Ministry of Health of the Russian Federation, 197376 Saint Petersburg, Russia
*Corresponding author: Irina Tcymbarevich, CJSC Biotechnology Developments, 119136 Moscow, Russia
Received Date: 18 July 2025
Accepted Date: 23 July 2025
Published Date: 25 July 2025
Citation: Kuldyushev N, Tcymbarevich I, Stukova M, Romanovskaya-Romanko E, Sukhova M, et al. (2025) Development and Initial Design of the Hemagglutinin Stem Region in a Universal Influenza Vaccine. Adv Biochem Biotechnol 10: 10125 https://doi.org/10.29011/2574-7258.010125.
Abstract
Objectives: Influenza is a socially and economically significant disease, which requires regular vaccination of the population due to the high variability of the virus.
Methods: In this work, we designed and obtained a recombinant protein - the stem part of influenza hemagglutinin, which has potential as an antigen in a universal vaccine against influenza virus. We also describe the development of a stable recombinant manufacturing platform based on Chinese Hamster Ovary (CHO) cells as a host for the target protein production. Genetic construct encoding a recombinant protein, a polyHis tag, and a fluorescent reporter was generated using standard genetic engineering methods.
Results and Conclusions: Two stable high-producing CHO cell pools clones exhibiting the highest levels of fluorescence intensity were sorted by FACS for further production of the recombinant protein. The protein was purified by immobilized metal affinity chromatography in sufficient quantities for subsequent functional tests. The purified recombinant protein can be further evaluated for its ability to elicit broadly neutralizing antibodies against the influenza virus.
Keywords: CHO; Genetic Engineering; Hemagglutinin, Influenza; Protein Production; Stem Part; Universal Vaccine; Vaccines
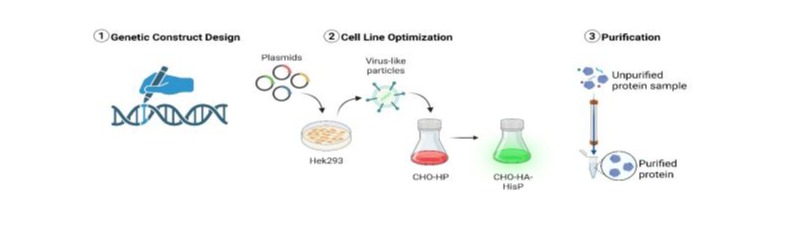
Introduction
Influenza represents a disease of substantial public health importance, imposing a significant burden on global healthcare systems, with an estimated annual mortality exceeding 500,000 cases worldwide [1]. Conventional inactivated influenza vaccines, predominantly produced in embryonated chicken eggs, exhibit notable limitations, including strain-specific efficacy and suboptimal protection against seasonal and pandemic variants [2]. Furthermore, this production method may introduce antigenic modifications that compromise vaccine immunogenicity [3]. Consequently, there is a compelling rationale for transitioning toward a universal influenza vaccine platform employing recombinant antigen(s) to overcome these constraints [3]. A promising strategy for universal vaccine development involves the utilization of the stem domain of the influenza surface glycoprotein Hemagglutinin (HA) as a target antigen. This region is highly conserved across influenza strains, eliciting broadly neutralizing antibodies with cross-reactive specificity against multiple subtypes within influenza A groups and, in certain cases, even crossgroup neutralization [4-9]. Notably, immunological studies have demonstrated differential B-cell responses to conserved HA stem epitopes following vaccination with H7N9 (group 2) or H5N1 (group 1) influenza strains. Intriguingly, individuals immunized with group 2 HA exhibit B-cell populations capable of recognizing HA stem epitopes from both group 1 and group 2 viruses, suggesting broader immunogenic potential [10]. These findings support the hypothesis that the HA stem domain from group 2 influenza viruses may serve as an optimal antigenic candidate for a recombinant universal influenza vaccine. The primary objective of the current study was to produce ≥500 µg of recombinant HA stem protein, both with and without a C-terminal poly-histidine tag. To achieve this, the following experimental tasks were delineated:
- Genetic Construct Design: Generation of an expression vector encoding the target protein, incorporating a C-terminal linker, poly-histidine tag, and fluorescent reporter for detection.
- Cell Line Optimization: Generation of at least two heterogeneous cultures of HEK or CHO cells exhibiting maximal fluorescence intensity, indicative of high recombinant protein expression.
- Protein Production and Purification: Establishment of a scalable protocol for the isolation and purification of the target molecule. Preparative purification yielding 300-500 µg of the recombinant HA stem protein, both in tagged (His-tagged) and untagged forms.
Materials and Methods
Plasmids and primers
P-Pharma Zeo RBD-Fc, KA0717_pPB-hCMV*1-cHA-IRESVenus (Addgene, USA), pLeGO-G2 expression vectors were used to assembly genetic construct. Primers were synthesized in house. Primer sequences are listed in the Supplement Table S1. Fragment sizes were verified by agarose gel electrophoresis, followed by gel extraction using Cleanup Mini kit (Eurogen, Russia).
Design and cloning of the coding sequence
All nucleotide fragments were amplified by PCR using a readyto-use reaction mixture with the high-fidelity Q5 polymerase from the Q5 Hot Start High-Fidelity 2X Master Mix kit (NEB, USA). PCR products were isolated from the reaction mixtures using magnetic beads KAPA Pure Beads (KAPA Biosystems, USA). All PCR results were confirmed by electrophoresis in a 1-1.5% agarose gels containing 0.05% ethidium bromide solution. DNA bands were visualized under UV light with Sky High DNA ladder (Biolabmix, Russia). Gene sequence was codon-optimized for CHO using GenScript (https://www.genscript.com/tools/ gensmart-codon-optimization) and purchased in Eurogen, Russia. The pLeGO- backbone fragment was amplified by PCR using linearized pLeGO vector as template (7,317 bp). The CMVenEFa1 fragment was generated by PCR amplification. PCR products were purified using KAPA Pure Beads. Fusion fragment CMVen-EFa1-HA-His-1 was generated by overlap extension
PCR. For generation of intermediate plasmid, CMVen-EFa1-HAHis fragment and pLeGO-backbone were assembled using Gibson Assembly [13]. The reaction mixture was transformed into E. coli NEB Stable cells to minimize plasmid recombination. One of the colonies contained the desired insert, with sequence accuracy confirmed by Sanger sequencing. The full-length expression vector was assembled using the previously constructed pLeGO -CMV-HAHis and IRES-mVenus fragment. pLeGO-CMV-HAHis was linearized by PCR and fragment with the correct size was extracted from agarose gel. IRES-mVenus fragment was amplified by PCR. Final vector assembly was performed using Gibson Assembly [13]. The reaction mixture was transformed into NEB
Stable competent cells. Positive clones were identified by two PCR screening strategies: primers 5 and 8 were used to confirm IRES and mVenus presence Table S1, Figure S1; primers 13 and 14 for EF1a promoter, target sequence, and IRES Table S1, Figure S1, respectively. The complete genetic sequence was validated by Sanger sequencing. The genetic sequences were validated by Sanger sequencing Supplement Figure S1. Plasmid DNA was prepared using a Maxi plasmid DNA isolation kit (Biolabmix, Russia). The obtained DNA was aseptically ethanol-precipitated following the manufacturer’s protocol.
|
No |
The list of primers |
|
Primer sequence (5’ → 3’) |
|
|
1 |
ggacagcagagatccagtttggttagtacctcttcacgctccattgcc |
|
4 |
atatcaagcttatcgggagctacgtacgttagtggtg |
|
5 |
aacgtacgtagctcccgataagcttgatatcgaattc |
|
7 |
cacgatgataatctggccacaaccatggt |
|
8 |
tgctatacgaagttattaggtccctcgacgcgctttacttgtacagctc |
|
13 |
cttggttcattctcaagcctcag |
|
14 |
caccccggtgaacagc |
|
15 |
ctgtacaagtaaagcgcgtcgaggga |
|
16 |
ggtactaaccaaactggatctctg |
|
21 |
gcagagtgtggctgtg |
|
22 |
gctgaaccagcacaccatc |
Supplement Table 1: Primer sequences.
Primer 7
Primer 13
Primer 14
Primer 21
Primer 22
Figure S1: Sanger Sequencing.
Cell Cultures
HEK293TN cells for lentiviral particles production were cultured in DMEM medium (PanEco, Russia) supplemented with 1 g/L glucose (PanEco, Russia) and 4 mM L-glutamine (PanEco, Russia). Chinese Hamster Ovary cells (CHO-S, Thermo Fisher Scientific, Monza, Italy) were cultured in EmCD CHO 104 medium (Eminence, China) supplemented with 4 mM L-glutamine (PanEco) kept at 37 °C on an orbital shaking CO2-incubator (120 rpm). Cells were cultured in Erlenmeyer flasks. Viability, pH, glucose concentration and cell concentration were measured once in 1-2 days. Cell counts and viable cells were measured after staining with Trypan Blue using a Countess 3 cell counter (Thermo Fisher Scientific).
Production of Viral Particles
Viral particle production was performed using HEK293TN cells. Cells were maintained in T-25 flasks until reaching 80% confluency. Transfection was conducted using the pLeGO-HA-His vector along with helper plasmids Md2G and PsPax2 encoding viral packaging genes, employing PEI as the transfection reagent. After 24 hours, the culture medium was replaced with fresh medium containing 1 mM sodium butyrate (Sigma-Aldrich, USA). On the third day after transfection, cell culture medium with lentiviral particles was collected, filtered with 0.45 µm membrane filters and centrifuged at 3,000 × g, 16 h, 4°C. The transparent precipitate was collected and reconstituted in 1 mL of CHO cell culture medium.
CHO Cell Pool Generation
One day before transduction, 100,000 cell were seeded in 2 ml of culture media. Cells were incubated with 125 μl of viral particle supernatant and 4 μg/ml protamine sulfate (Ferain OJSC, Russia). Three days post-transduction, cells were harvested by centrifugation (120 × g, 5 min) and resuspended in fresh medium at 5*10⁶ cells/mL. Transduced cells were cultured in a 5% CO₂ incubator for 3 days prior to sorting.
Cell sorting
Cell sorting was performed using a BD FACSAria III cell sorter equipped with: Aerosol Management System (AMS), 130 μm nozzle, 495 nm laser excitation. Cells were gated into GFPMed (medium fluorescence) and GFP-Hi (high fluorescence) populations.
Protein Expression
For protein expression, cells were seeded in 125 ml of CHO culture medium with 0.1% anti-clumping agent in two 500 mL Erlenmeyer flasks with starting density 106 cells per ml. Glucose levels were measured using a Satellite Express glucometer (Russia). pH was measured using pHScan indicator strips (pH 5.4-10.0, pHSCAN, Russia). The feed supplements used were EmCD CHO 101 Feed A and EmCD CHO 101 Feed B (Eminence, China). Cultivation was stopped when cell viability dropped below 85%. pH was maintained at 7.0±0.2. After cultivation, the culture medium was clarified in two steps. The 1st step involved centrifugation at 300 g for 10 minutes at 4°C. In the 2nd step, the supernatant was transferred to clean tubes and centrifuged at 3000 g for 10 minutes at 4°C. The supernatant was then transferred to clean tubes and filtered using 0.45 µm PES membrane filters. For sterilization, the culture medium was filtered through 0.22 µm PES membrane filters.
Protein Purification and SDS-PAGE
Initial dialysis of the clarified cell medium was performed using tangential flow filtration with a Sartorius VivaFlow 50 cassette (5 kDa MWCO). The system was prepared through sequential washing with MilliQ water, 0.5 M NaOH sanitization, and final equilibration with binding buffer (50 mM Tris, 300 mM NaCl, 5 mM imidazole, pH 7.5). The purification protocol employed immobilized metal affinity chromatography (IMAC) using an ÄKTA Avant 150 system with Nuvia IMAC resin. Following column equilibration, the sample was loaded and subjected to washing with imidazole-containing buffer. Elution of the target protein was achieved using 250 mM imidazole buffer, yielding 8 mL of eluate. Eluate was concentrated and buffer exchanged steps were performed using Vivaspin 6 centrifugal concentrators (30 kDa MWCO). Denaturing polyacrylamide gel electrophoresis (SDSPAGE) was performed to confirm the presence of the protein. 12% PAGs were prepared with 30% acrylamide/bis solution (29:1) in 375 mM Tris and 0.1% SDS. The electrophoresis was performed with 1X Tris-Glycine running buffer (25 mM Tris, 250 mM glycine, 0.1% SDS). Resolved proteins were visualized by 30 min of Coomassie Brilliant Blue R-250 (Bio-Rad, USA).
Results
Construction of a Genetic Vector with C-Terminal Linker and Poly-histidine Tag
Protein Sequence Selection
Several engineered stabilized variants of H3 and H7 hemagglutinin stem domains have been previously described in the literature [11]. The H3ssF_C sequence was selected for this study based on three key characteristics: (1) demonstrated structural stability, (2) recognition by all tested broadly neutralizing antibodies, and (3) moderate expression yield in recombinant systems. The H3ssF_C protein represents a modified version of influenza H3N2 (A/ Finland/486/2004) HA with the globular head domain removed and containing specific mutations to preserve proper protein folding, structural integrity, and immunogenicity Table 1.
|
Side-chain substitutions to restore the globule (H3 numbering) |
C-terminal extension of helix A |
N-terminal extension of helix C |
Capping of helix C |
β-hairpin |
Helical linker |
S-S bond |
|
K51HA2M, L52HA2V, S93HA2A, N95HA2L, E103HA2L, T107HA2V, N116HA2R |
ELMEQ
|
NA
|
D
|
VFPGCGV
|
GGPD
|
42eHA1-93HA2
|
Table 1: Nucleotide substitutions
The target protein sequence was derived from the published H3ssF_C construct [11], with the following additions: (1) a C-terminal hexahistidine tag (His tag) to enable immobilized metal affinity chromatography purification from culture supernatant, (2) a Tobacco Etch Virus (TEV) protease recognition sequence inserted between the H3ssF_C sequence and poly-histidine tag to allow tag removal, and (3) an intervening linker sequence to prevent steric hindrance during either proteolytic cleavage or purification procedures. The complete amino acid sequence of the final construct HA-His is presented in Figure 1A.
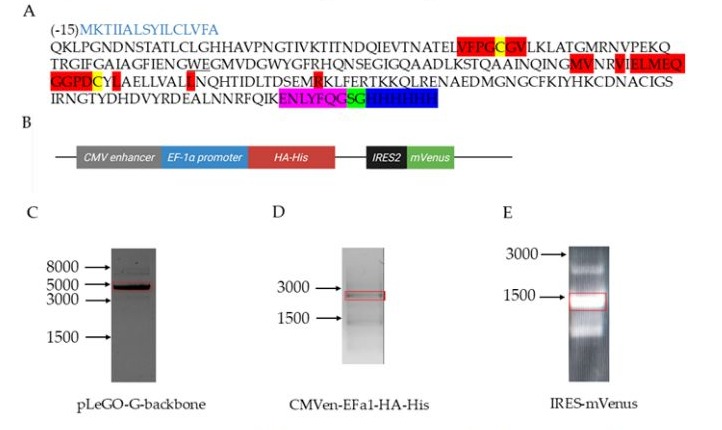
Figure 1: Sequence of the target recombinant protein HA-His - the stem part of influenza HA. (A) Amino acid sequence. The substitutions are highlighted in red; cysteines are highlighted in yellow, the TEV protease recognition site is highlighted in pink, the linker is highlighted in green, and the poly-His tag is highlighted in blue. The peptide signal sequence is marked in light blue font, and the amino acids W95, E96, G97 are underlined.
(B)Fragment of the plasmid map. The structural elements are labeled as follows: CMV enhancer - CytoMegaloVirus enhancer; EF1a promoter - human EF1a promoter; HA-His - sequence encoding the target protein; IRES2 - internal ribosome entry site enabling capindependent translation initiation; mVenus - sequence encoding the fluorescent marker mVenus. (C)Agarose electrophoresis of pLeGObackbone fragment (red rectangle). PCR was run with primers 15 and 16 Table S1. (D)Agarose electrophoresis of CMVen-EFa1-HAHis fragment (red rectangle). PCR was run with primers 1 and 4 Table S1. (E)Agarose Electrophoresis of IRES-mVenus fragment (red rectangle). PCR was run with primers 5 and 8 Table S1. Arrows indicate DNA ladder/b.p.
Generation of the Target Protein Expression Vector
The amino acid sequence was converted to a nucleotide sequence with codon usage optimized primarily for Cricetulus griseus (CHO) cells and secondarily for human cell expression systems. The resulting nucleotide sequence was analyzed for the presence of strong ribosome binding sites (GGAGG and TAAGGAG). The analysis revealed a strong GGAGG ribosome binding site within the sequence TGGGAGGGA, corresponding to codons for residues W95, E96, and G97 (H3ssF_C numbering; Figure 1A. In CHO cells, tryptophan (W) is encoded exclusively by TGG, glutamate (E) by either GAG or GAA, and glycine (G) by any GGN codon. Therefore, elimination of the undesirable GGAGG sequence could only be achieved by modifying the E96 codon. Additionally, the sequence was verified to lack potential transcriptional terminators (poly-A: AAAAA; poly-T: TTTTT). The nucleotide sequence encoding the target protein is presented in the Supplement Table S2.
|
Sequence (5’ → 3’) |
|
atgaagaccatcatcgctctgagctacatcctgtgtctggtcttcgcccagaagctgcctggcaacgacaacagcacagccacactctgcctgggccatcacgccgtgcccaacggcaccatcgtgaagaccatcaccaatgatcagatcgaagtgaccaacgctaccgagctggtgttccctggctgtggcgtgctgaaactggctaccggcatgagaaacgtgcctgagaagcagacaagaggcatcttcggcgccattgccggcttcatcgagaatggctgggaaggaatggtggacggctggtatggctttagacaccagaactctgaaggcatcggccaggctgccgatctgaagtccacacaggccgctatcaaccagatcaacggtatggttaacagagtgatcgaactgatggaacaaggcggcccagattgctacctggccgaactgctggtggccctgctgaaccagcacaccatcgatctgaccgacagcgagatgcggaagctgttcgagagaaccaagaagcagctgagagagaacgccgaggacatgggcaatggatgcttcaagatctaccacaagtgcgacaacgcctgcatcggatccatccggaacggtacctacgaccacgacgtgtaccgggacgaggctctgaacaaccggtttcaaatcaaagagaacctgtacttccagggctctggccaccatcaccaccaccac |
Supplement Table 2: Nucleotide sequence of the target protein.
The expression cassette consisted of the following elements Figure 1B: CytoMegaloVirus (CMV) enhancer and human EF1α promoter, target protein coding sequence, IRESv7, and fluorescent reporter mVenus. The construct features a Kozak sequence (TAGCCGCCACC) upstream of the start codon. The IRES element was cloned from plasmid KA0717_pPB-hCMV*1-cHA-IRESVenus and modified by introducing a 3’ nucleotide substitution (IRESv7 as described in [12], which according to literature reduces the downstream gene expression by 3-fold compared to the wild-type [12]. This is followed by the mVenus sequence, selected for its monomeric nature and superior brightness compared to the original GFP in the pLeGO vector. The coding sequence was cloned into a lentiviral expression vector pLeGO in two steps. First, an intermediate plasmid containing CytoMegaloVirus (CMV) enhancer and human EF1α promoter along with the hemagglutinin stem and poly-histidine tag coding sequences was cloned into pLeGO-G2 backbone. Second, capindependent ribosome binding site (IRES) IRESv7 and yellow fluorescent protein mVenus nucleotide sequences were cloned into the intermediate plasmid. All fragments were amplified with high-fidelity polymerases Figure 1C, D, E and cloned using Gibson assembly to allow scar-less joining.
Generation of Heterogenous Cell Cultures
Successfully produced lentiviral particles were used for transduction of CHO-HP cells. Since genetic construct had IRES between HAHis and mVenus sequences, all fluorescent cells should express HA-His. To isolate cells expressing HA-His, we sorted cells with the highest fluorescence intensity. Single-cell sorting gates (Singl_FCS, Singl_SSC) were established using non-transduced control cells based on forward and side scatter profiles Figure 2A. Transduced cells were gated into GFP-Med (Medium Fluorescence) and GFP-Hi (High Fluorescence) populations based on mVenus expression Figure 2B, representing 45% and 35% of total viable cells, respectively. Cell pool in GFP-Hi population was called CHO-HO-HA-His0. After biomass expansion, these cells were re-sorted to exclude nonfluorescent cells. The final heterogeneous cell pool is called CHO-HP-HisP. These cells are extremely bright meaning that they have high expression of HA-His, at least, on the mRNA level.
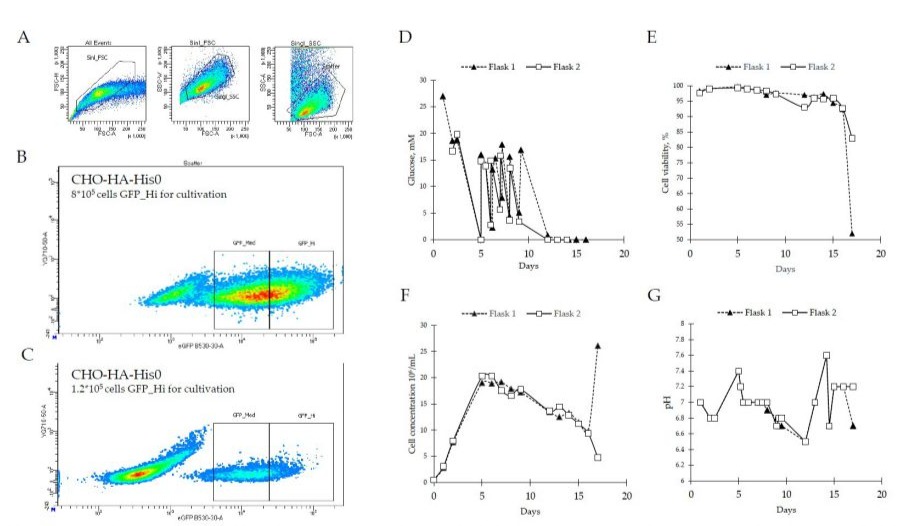
Figure 2: Development of stable CHO cell system for production of HA-His. (A) Gating strategy. Single-cell sorting gates (Singl_FCS, Singl_SSC) were defined using non-transduced control cells according to forward and side scatter profiles. Transduced cells were subsequently sorted into GFP-Med (medium fluorescence) and GFP-Hi (High Fluorescence) subsets based on mVenus expression levels. (B) 1.2*105 CHO cells with high expression of mVenus were sorted to obtain the 1st heterogenous CHO strain - CHO-HA-His0. (C) CHO-HA-His0 were 8*105 cells with high expression of mVenus were sorted to obtain the 2nd heterogenous CHO strain - CHO-HAHisP, which were used for further cultivation. CHO-HA-HisP cell growth characteristics were: (D) Glucose concentration, mM; (E) Cell viability, %; (F) Cell concentration, 106/m; (G) pH. Abbreviations: FSC-H - Forward scatter height; FSC-A - Forward scatter area; SSC-W - Side scatter width; SSC-A - Side scatter; YG710-50-A - Area under the curve (excitation 561 nm, detector 710/50 nm); eGFP B530-30-A - GFP channel (excitation 488 nm, detector 530/30 nm).
Fed-Batch Cultivation
The CHO-HA-HisP cell pool was selected for protein production because it had cells expressing higher level of the fluorescent marker mVenus. Since the marker is expressed from the same mRNA as the target protein (HA), the protein yield was expected to be higher as well. To produce HA-His in sufficient amounts, we performed fed-batch cultivation in flasks for 17 days. Starting from the second day of cultivation, we monitored glucose levels, cell viability, cell concentrations and pH Figure 2D, E, F, G. To potentially increase protein yield, the cultivation temperature was reduced to 35°C on the 2nd day and further to 33°C on the 5th day of cultivation. Cultivation was stopped when cell viability dropped below 85%. To maintain pH at 7.0±0.2 we adjusted % CO2 and modified the feed volume once CO2 was set 0% Figure 2G. After cultivation, the culture medium was clarified in two steps to remove cells and to remove cell debris. The supernatant was filter-sterilized in two steps with 0.45 µm and 0.22 µm PES membrane filters. The resulting clarified culture medium was used for purification.
Protein Purification
The purification process with clarified cell culture supernatant containing 0.1% anti-aggregation reagent required removal prior to chromatography. Initial dialysis was performed using tangential flow filtration with 5 kDa molecular weight cutoff equilibrated with binding buffer. The purification protocol employed immobilized metal affinity chromatography. Following column equilibration, the sample was loaded and subjected to washing with imidazole-containing buffer Figure 3A. Elution of the target protein was achieved using 250 mM imidazole buffer, yielding 8 mL of eluate. HA-His is expected to have trimeric state and mass more than 78 kDa, thus we used centrifugal concentrators with molecular weight cutoff 30 kDa. After concentration and buffer exchange yield was 4.2 mg of purified His-tagged protein as quantified by UV spectrophotometry.
Figure 3: Protein purification. Chromatography and 12-% Tris-Glycine SDS-PAGE under denaturing conditions. Collected fraction is marked with red; Absorbance at 280 nm - blue, 260 nm - orange; Imidazole gradient - black. Estimated calculated M.W. ~27 kDa. (A)
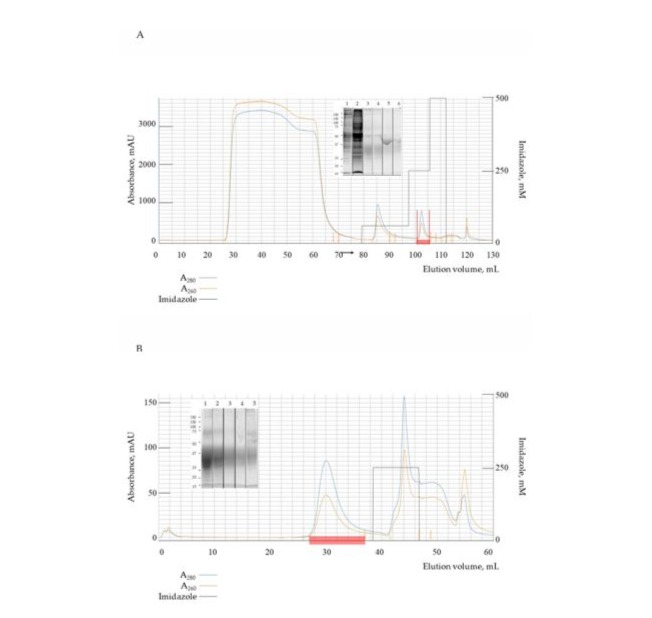
Chromatography profile of clarified culture medium. Lane 1 - loaded sample. Lane 2 - washed sample. Lane 3 - undiluted eluate. Lane 4 - 1:2 eluate dilution. Lane 5 - 1:5 eluate dilution. Lane 6 - 1:10 eluate dilution. Estimated calculated M.W. ~27 kDa; (B) Chromatography profile of target protein after His-tag removal. Lane 1 - undiluted eluate. Lane 2 - 1:2 eluate dilution. Lane 3 - 1:5 eluate dilution. Lane 4 - 1:10 eluate dilution. Lane 5 - rinsed column. Estimated calculated M.W. ~26 kDa.
For tag removal, the construct’s TEV protease recognition site was utilized to cleave using His-tagged TEV protease. The tagless protein was then separated from uncleaved product and protease through a second metal affinity chromatography step, yielding 1.4 mg of
purified protein Figure 3B. Process efficiency and product purity were verified by SDS-PAGE analysis for both chromatography steps.
Discussion and Conclusions
This work successfully established the production and purification protocol for the stem region protein of a universal influenza vaccine candidate, achieving both tagged and untagged variants suitable for further characterization and immunological studies. The methodology demonstrates robust protein recovery while maintaining structural integrity, as evidenced by the consistent performance across multiple purification steps. These results represent a significant step toward developing a broadly protective influenza vaccine based on the conserved hemagglutinin stem domain. The differential B-cell responses elicited by group 2 (H7N9) and group 1 (H5N1) influenza strains highlight the importance of HA stem immunogenicity in universal vaccine design. Immunological studies demonstrate that group 2 HA vaccination induces broader cross-reactive B-cell responses against conserved stem epitopes compared to group 1 strains, which tend to elicit more subtype-restricted immunity [14,15]. This dichotomy may stem from structural differences in group 1 and group 2 HA stems, particularly in glycosylation patterns and conformational flexibility, which influence antibody accessibility [16, 17]. Notably, group 2 HA stems appear to preferentially activate B-cell clones capable of recognizing shared epitopes across both groups, suggesting their superior potential as universal vaccine antigens.
The conserved nature of HA stem epitopes across diverse influenza strains, including pandemic H1N1 and seasonal H3N2 variants, further supports their targeting for broad protection [18,19]. However, pre-existing immunity from prior exposures can modulate these responses, with repeated vaccinations sometimes limiting de novo B-cell activation against stem epitopes [20]. Our successful purification of a stable, recombinant HA stem protein provides a tool to dissect these responses systematically. Future studies should evaluate its ability to recapitulate the cross-reactive B-cell activation patterns observed with natural group 2 infections and assess its immunogenicity. These insights could guide the development of multi-component universal vaccines capable of eliciting robust, long-lasting immunity against diverse influenza strains. Future studies will include sequential and combined immunization schemes to investigate the best strategy to elicit associated serum antibody responses.
Supplementary Materials: Figure S1: Sanger sequencing; Table S1: The list of primers.
Author Contributions: N.K. and I.T. contributed equally to the manuscript. Conceptualization, funding acquisition, I.K.; writing—original draft preparation, I.T, N.K.; visualization, I.T, M.S.; investigation, supervision, project administration, writing - editing, N.K. Methodology and, investigation, M.St, E.R.-R. All authors have read and agreed to the published version of the manuscript.
Funding: This research was funded by CJSC Biotechnology Developments. This research received no external funding.
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: The original contributions presented in this study are included in the article/supplementary material.
Further inquiries can be directed to the corresponding authors.
Acknowledgments: Graphical abstract was created with the help of BioRender.com
Conflicts of Interest: The authors declare no conflicts of interest.
References
- Fischer II W.A, Gong M, Bhagwanjee S, Sevransky J. (2014) Global Burden of Influenza as a Cause of Cardiopulmonary Morbidity and Mortality. Global Heart 9 :325-336.
- CDC Seasonal Flu Vaccine Effectiveness Studies (url: https://www. cdc.gov/flu-vaccines-work/php/effectiveness-studies/index.html, accessed on 10.04.2025).
- Paules C.I, Sullivan S.G, Subbarao K, Fauci A.S. (2018) Chasing Seasonal Influenza. The Need for a Universal Influenza Vaccine. N Engl J Med.378: 7-9.
- Okuno Y, Isegawa Y, Sasao F, Ueda S. (1993) A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J Virol. 67:2552-2558.
- Ekiert D.C, Bhabha G, Elsliger M.-A, Friesen R.H.E, Jongeneelen M. et al. (2009). Antibody Recognition of a Highly Conserved Influenza Virus Epitope. Science 324: 246-251.
- Sui J, Hwang W.C, Perez S, Wei G, Aird D, et al. (2009) Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 16:265-273.
- Corti D, Voss J, Gamblin S.J, Codoni G, Macagno A, et al. (2011) A Neutralizing Antibody Selected from Plasma Cells That Binds to Group 1 and Group 2 Influenza A Hemagglutinins. Science333: 850-856.
- Dreyfus C, Laursen N.S, Kwaks T, Zuijdgeest D, Khayat R, et al. (2012) Highly Conserved Protective Epitopes on Influenza B Viruses. Science 337: 1343-1348.
- Joyce M.G, Wheatley A.K, Thomas P.V, Chuang G.-Y, Soto et al. (2016) Vaccine-Induced Antibodies that Neutralize Group 1 and Group 2 Influenza A Viruses. Cell. 166: 609-623.
- Andrews S.F, Joyce M.G, Chambers M.J, Gillespie R.A, Kanekiyo M, et al. (2017) Preferential induction of cross-group influenza A hemagglutinin stem-specific memory B cells after H7N9 immunization in humans. Scie Immunol 2:13.
- Corbett K.S, Moin S.M, Yassine H.M, Cagigi A, Kanekiyo M, et al. (2019) Design of Nanoparticulate Group 2 Influenza Virus Hemagglutinin Stem Antigens That Activate Unmutated Ancestor B Cell Receptors of Broadly Neutralizing Antibody Lineages. mBio 10: 1.
- Koh E.Y.C, Ho S.C.L, Mariati, Song Z, Bi X,et al. (2013) An Internal Ribosome Entry Site (IRES) Mutant Library for Tuning Expression Level of Multiple Genes in Mammalian Cells. PLoS ONE. 8:12.
- Gibson D.G, Young L, Chuang R.-Y, Venter J.C, Hutchison C.A, et al. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 6:343-345
- Andrews, S. F, Graham, B. S, Mascola, J. R, McDermott, A. B. (2018). Is it possible to develop a “universal” influenza virus vaccine? Immunogenetic considerations underlying B-cell biology in the development of a pan-subtype influenza A vaccine targeting the hemagglutinin stem. Cold Spring Harb Perspect Biol, 10.
- Joyce MG, Wheatley AK, Thomas PV, Chuang GY, Soto C, et al. (2016) Vaccine-Induced Antibodies that Neutralize Group 1 and Group 2 Influenza A Viruses. Cell. 166:609-623.
- Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, (2011) A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science.333: 850-856.
- Yassine HM, Boyington JC, McTamney PM, Wei CJ, Kanekiyo M, et al. (2015) Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat Med. 21:1065-1070.
- Impagliazzo A, Milder F, Kuipers H, Wagner MV, Zhu X, et al. (2015) A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science. 349:1301-1306.
- Krammer F. (2019) The human antibody response to influenza A virus infection and vaccination. Nat Rev Immunol. 9: 383-397.
- Skowronski DM, Chambers C, Sabaiduc S, De Serres G, Winter AL, et al. (2016) A Perfect Storm: Impact of Genomic Variation and Serial Vaccination on Low Influenza Vaccine Effectiveness During the 20142015 linical infectious diseases : an official publication of the Infectious, Diseases Society of America, 63:21-32.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.