Complete Rhabdomyoblastic Differentiation in a Leptomeningeal Metastatic Drop of Medulloblastoma- Case Report
by Maryam Almurshed1*, Fatma Dashti2, Nisreen Khalifa3, Radovan Mijalcic4
1Department of Pathology, Al Sabah Hospital, Ministry of Health, Kuwait
2Department of Radiology, Ibn Sina Hospital, Ministry of Health, Kuwait
3Department of Pediatric Oncology, NBK hospital, Ministry of Health, Kuwait
4Department of Neurosurgery, Ibn Sina Hospital, Ministry of Health, Kuwait
Received Date: October 12, 2025
Accepted Date: October 24, 2025
Published Date: October 27, 2025
Citation: Almurshed M, Dashti F, Khalifa N, Mijalcic R (2025) Complete Rhabdomyoblastic Differentiation in a Leptomeningeal Metastatic Drop of Medulloblastoma- Case Report. J Neurol Exp Neural Sci 7: 166. https://doi.org/10.29011/2577-1442.100066.
Abstract
Background: Medulloblastoma with divergent differentiation is a rare event, which was investigated in small prior series, however, lumped under the molecular subgroups nowadays. Studing their disease progression can give insight to tumor response to current therapy. Case Presentation: Four years old boy presented with cerebellar mass, that is diagnosed as Medulloblastoma, desmoplastic/nodular morphology with MET point mutation, who relapsed as spinal leptomeningeal disease after compoletion of his chemo- and radiotherapy in the form of complete rhabdomyoblastic nodule -by histopathology and immunohistochemistry- at T12/L1, and later along the course of disease in T3-T4 location. The metastatic deposit had no neuronal, medulloblastoma, neuropil, small round blue cell differetiation. Later the patient disease progressed while on 2nd line of therapy, developed paraplegia and died of disease within 3 years of presentation. Conclusion: Medulloblastoma has been known to be able rarely to develop myogenic differetiation, however, detailed morphology of either smooth muscle or skeletal differentiation in literature is lacking. In addition, the metastatic progression of disease was always assumed to be of the same morphology of the primary, because most likely it is not biopsied, however, this case proves that it can have different morphologies other than the primary, which is even rarer -not even reported- of rhabdomyoblastic nodule- as transdifferentiation of the medulloblastoma. This transdiffrentiation can have an impact on patient response to future therapy and subsequently disease progression and patient outcome.
Keywords: Medulloblastoma; Myogenic differentiation; Case report; Forth ventricle
Abbreviations: CNS: Central Nervous System; GCS: Glascow Coma Scale; HS: Histiocytic Sarcoma; RT: Radiotherapy; CT: Chemotherapy; MRI: Magnetic Resonance Imaging; FISH: Fluorescent Insitu Hybridization; BBB: Blood brain barrier; ALCL: Anaplastic large cell lymphoma; CT: Computed tomography; NF: Neurofilament
Introduction
Medulloblastoma with divergent differentiation is a rare event. The reported patterns of differentiation include glial, melanocytic, myogenic.41,44 The effect of differentiation on tumor behavior is not well understood, as nowadays, they have been lumped under their molecular groups for expected behaviour and theraputic management issues.4, 41 Rare studies have adopted studing their disease progression and patient prognosis.28,40 However, rarely the relapse gets biopsied to assess the response/resistance to therapy on the relapsed focus- as in other organs. For that we present this interesting case, that might guide us for future managements in medulloblastoma cases.
Case Presentation
Four years old boy who presented primarily with history of vomiting for 2 weeks followed by headache of gradual onset and progressive in coarse, where he was managed by supportive therapy without improvement. Afterwards the patient symptoms progressed to ataxia, somnolence, anorexia and developed squint, however, there is no symptoms of fever, night sweat or weight loss. The child is a product of consanguineous marriage and known for G6PD. After hospital admission, upon physical examination the patient was agitated, vitally stable, with normal body systematic examination, except for the CNS. The patient revealed convergent squint bilaterally, and mutism.
Patient work-up and Investigation
C.T scan was done that revealed hydrocephalus and posterior fossa mass. Subsequent MRI brain showed a large 4th ventricular mass that was predominantly solid with multiple cystic foci, measuring 5 x 4 cm. It exhibited heterogeneous T2 hyperintensity with diffusion restriction and heterogeneous post-contrast enhancement (Figure 1). There were foci of susceptibility on SWI, of calcification and microhemorrhage. There was associated mass effect on the adjacent posterior fossa structures with bilateral cerebellar tonsillar herniation and secondary moderate supratentorial hydrocephalus. There was no other supra or infra tentorial parenchymal or meningeal lesions and no spinal drop metastasis. An EVD tube was inserted through the right frontal approach and steroids were added. Suboccipital craniotomy was performed with total excision of the mass.
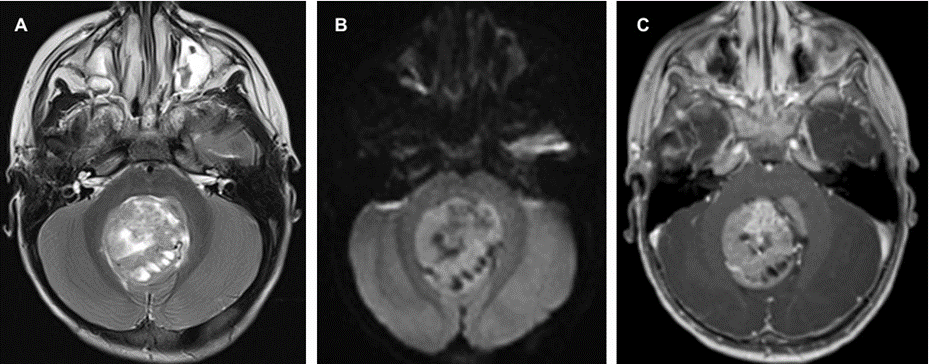
Figure 1: Preoperative MRI demonstrating the posterior fossa mass of heterogenous solid and cystic components (a) diffusion restriction on DWI (b) and heterogenous enhancement (c).
Histopathology, Immunohistochemistry and Molecular Studies
Histological sections show infiltrative tumor with nodular architecture, highlighted by Reticulin stain. The nodal areas are composed of mature neuronal elements (Figure 2). The internodal zone consists of undifferentiated cells with high N:C ratio and moderate atypia and pleomorphism. Frequent mitosis, including atypical forms, necrosis and apoptosis are present. The tumor is positive for synaptophysin, GAB1, YAP1, while negative for GFAP, P53, B-catenin (cytoplasmic) and EMA. INI1 is retained. Rare strap cells noted and highlighted by desmin and myogenin immunostains. Case reported as “Medulloblastoma, Desmoplastic/Nodular histological variant, GAB1 positive, b-catenin and P53 negative. Subsequent molecular testing by Oncomine Comprehensive Assay that revealed MET: c.3029C>T, p.(T10101) with no rearrangements detected. No mutation detected in CTNNB1, PITCH1/2, TP53, SMO, or NMYC.
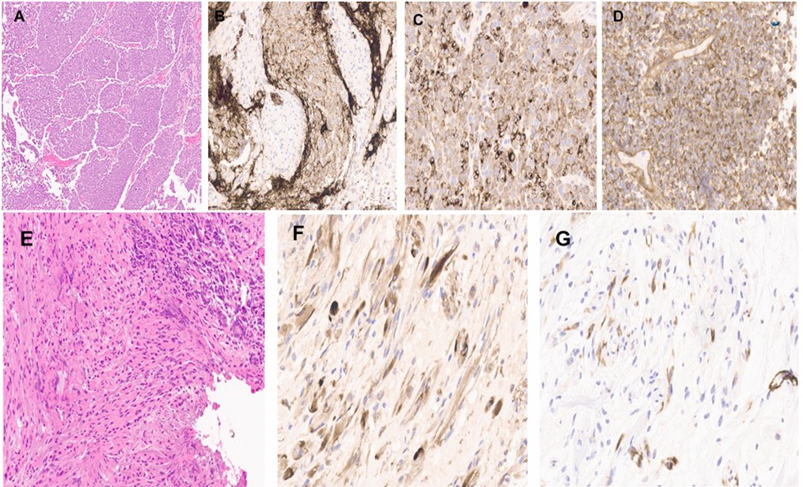
Figure 2: Low power view show the nodular growth pattern of cerebellar medulloblastoma (A). The tumor is positive for synaptophysin (B), NF (C), and GAB1 (D). Foci of spindle/strap cell differentiation (E). This component is positive for NF (F), and SMA (G).
Patient outcome and follow-up
Post op brain MRI brain revealed improvement of the hydrocephalous and complete surgical resection of the mass with associated post-operative parenchymal changes and residual blood products within the operative cavity. Patient received 6 cycles of chemotherapy according of PNET 5 Protocol, consisting of 3 courses of cisplatin, CCNU and vincristine alternating with 3 courses of cyclophosphamide and vincristine. He also received radiotherapy as per standard risk protocol, with total of 55.8 Gy. The therapy was complicated by multiple episodes of febrile neutropenia. On regular follow up MRI scan 23 months post-resection, a solid enhancing lesion was noted along the cauda equina, to the right of the conus medullaris at T12-L1 (Figure 3), which was excised.
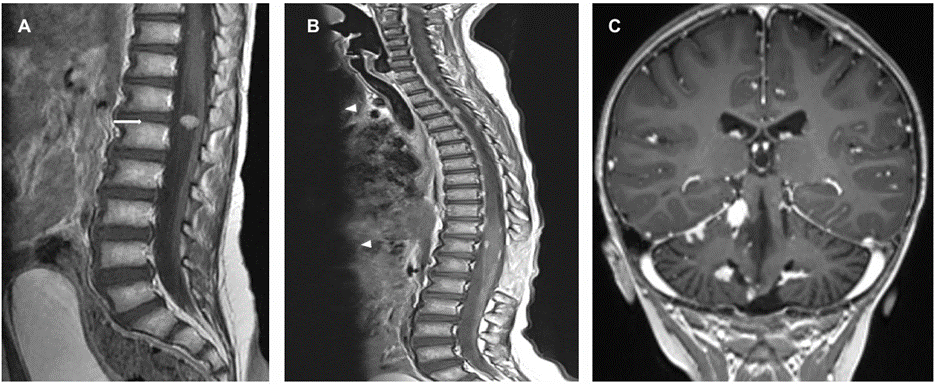
Figure 3: (a) Enhanced MRI spine showing the enhancing mass at the cauda equina (arrow). Follow up enhanced MRI of the spine (b) demonstrating the extramedullary enhancing lesions along the spine (arrowhead). (c) Coronal image along the cerebellum on the same MRI scan showing multiple enhancing lesions in the leptomeningeal distribution.
Histopathology, Immunohistochemistry and Molecular Studies
The excised mass was 1.5x1.0x0.3cm in dimensions and consists of a well circumscribed tumor, growing as intersecting fascicles, and vague wavy growth pattern. The tumor cells are monomorphic and show eccentric hyperchromatic nuclei with inconspicuous nucleoli, and eosinophilic cytoplasm with striation, as in mature rhabdomyoblastic differentiation (Figure 4). The mass lacks any mitosis, necrosis, round cell or dedifferentiated components. No evidence of medulloblastoma morphologically, neuropil or small round blue cell morphology.
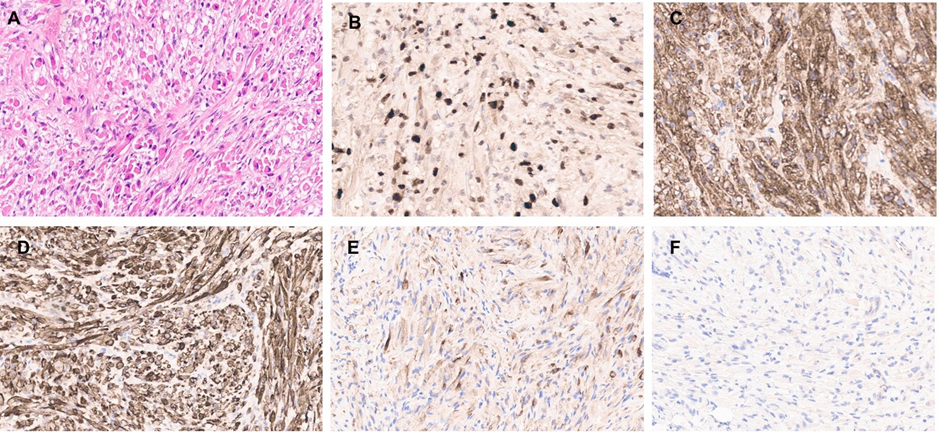
Figure 4: High power view of the spinal mass showing infiltration by rhabdoid-like cell (A). The cells are diffusely positive for myogenin (B), SMA (C), and Desmin (D). NF show scattered positivity (E). The tumour is negative for synaptophysin (F).
The cells stain positive for myogenic markers (myogenin, desmin), Neurofilament, while negative for glial markers (GFAP, Olig2), CD34, synaptophysin, and S100. INI1 and K27me are retained. The mass was sent for Oncomine Comprehensive Assay that revealed no clinically significant mutations or copy number variants detected. Oncomine comprehensive RNA assay did not detect any rearrangements. Further testing for Archer Fusion Plex Sarcoma Panel reveals absence of rearrangements.
Four months later, other extramedullary spinal lesions also appeared on MRI along the spinal cord as well as multiple intracranial posterior fossa enhancing lesions in leptomeningeal distribution (Figure 3); the largest measured 1.3 cm. A spinal lesion was later excised to show similar pathology. The case released as “Drop metastasis of Medulloblastoma with complete myogenic differentiation”. Afterwards, there was interval progression in the size and distribution of the posterior fossa and spinal leptomeningeal metastasis. The patient became paraplegic and was started on second-line chemotherapy, however, the disease progressed while on therapy and the patient died of disease 30 months after his primary presentation.
Discussion
Medulloblastoma, First described by Bailey and Cushing in 1925 [1]. as a rare brain embryonal tumor that grows from the fourth ventricle with a propensity to spread to the cerebellar vermis and the brainstem, seeding to the craniospinal axis through the subarachnoid system (the so-called “drop metastases” [2,3]. It is the second most common of all pediatric central nervous system tumors, accounting for about 20% of the total CNS tumors in the pediatric age group [3-5]. Disease relapse and metastasis is documented in 20-30% of medulloblastoma cases [6]. According to a systematic review, disease progression is through leptomeningeal dissamination or metastasis. The predominant sites of systemic metastases were bone -spine-, bone marrow and much rarer, metastases to soft tissues, lymph nodes, or lungs [7-11]. Leptomeningeal disease (LMD) is the presence of tumor cells in the leptomeninges, namely arachnoid and pia mater, or within the subarachnoid space into the CSF [6]. It is believed that as metastatic cells leave the primary tumor site, infiltrate the meninges, and spread via the arachnoid space or the CSF to form a metastatic colony, the MB cells are also discovered in the bloodstream of patients, which would suggest that LMD might arise due to the spread of circulating tumor cells through the blood [12,13]. NOTCH1 signalling pathway regulates both the initiation of metastasis and the self-renewal of medulloblastoma cells [14]. The molecular process in which MB cells acquire abilities to cause LMD likely involves genetic or epigenetic changes that guide Epithelial-to-mesenchymal transition (EMT) which allow the tumor to invade meningeal or endothelial barriers [15]. These changes help the cells to stay dormant and thereby resilient to radiation and chemotherapies. Afterwards, mesenchymal cancer cells that disseminated and formed new colonies concomitantly revert back to an epithelial phenotype in a mesenchymal-to-epithelial transition (MET) which is suggested to happen also in MB [16]. Another route of dissamination is surgical interventions and craniotomy which both interrupt the blood-brain barrier mechanically which enable the immigration of the tumor cells. Lymphatic and hematogenic (including the retroauricular and cervical lymph nodes) extensions of the primary tumor of the brain have already been proposed [9,10]. Another often proposed mechanism for the extra-neural extension of the medulloblastoma is iatrogenic diffusion through ventriculo-peritoneal shunts, which are primarily likely to result in peritoneal metastases.10 As high expression of CCL2 cytokine in CSF in metastastaic Group 3 and 4 MB, disease monitoring to rule out relapse is advised [17,18].
The presence of leptomeningeal disease (LMD) is higher in medulloblastoma relapse cases suggesting that metastasis highly linked to tumor progression and relapse, at least in non-WNT subgroup, as evidenced by the 70% of relapsed medulloblastomas with LMD [19]. Among medulloblastoma subgroups, GROUP 3 tumors show the highest rate of LMD and metastatic dissamination at presentation, followed by Group 4, SHH, and the least is WNT group, reported as >55%, 40%, 20% and 10% respectively.13 Those with MYC amplification of Group 3 has a higher risk than those without [20, 21]. and among SHH-activated group, than those with TP53 mutation show a higher degree of LMD compared to their TP53 wild-type counterparts [22, 23]. The molecular subgroup of the relapse remains the same as the primary tumor [24,25]. While SHH MBs show significantly more local relapses in the area where they originally arose (cerebellum), Group 3 and 4 MBs usually present with distant relapses or leptomeningeal spread [19]. The metastatic relapse of Group 3 MB is mostly multifocal or laminar whereas of Group 4 MBs present with isolated deposits. Compared to our present case, the tumor was of Desmoplastic/ nodular morphology, with MET point mutation, p53 wildtype, that relapsed as LMD, which is -according to literature- one of the least likely to dissaminate.
Medullomyoblastoma, first described by Marinesco and Goldstein in 1933 [26]. is an extremely rare clinical-pathological entity [27]. Non-WHO designated histologic variant of Medulloblastoma can present with myogenic differentiation, which is an aggressive malignant neoplasm seen predominantly in children. The tumor most commonly originates from the cerebellar vermis, although other sites include the cerebellar hemispheres [28] and the cerebellopontine angle [29,30]. Many hypothses about its histogenesis have been reported. Briefly, current hypotheses include MMD arising as part of a malignant teratoma [31,32] myoblastic differentiation of primitive neuroectodermal cells [33,34] and secondary myoblastic differentiation of mesenchymal or endothelial cells near or within a primitive neuroectodermal tumor [35,36] The disease have an extended morphological spectrum, that spans the presence of smooth muscle differentiation as smooth muscle actin positive cross-striated eosinophilic cells consistent with leiomyoblasts to rhabdomyoblastic differentiation. The prior does not seem to be associated with an improved outcome [37-39].
In literature, comprehensive description of the morphologic findings of medullomyoblastoma is lacking. To date, all reported tumors have been biphasic, containing primitive neuroectodermal and rhabdomyoblastic components as by definition, however, rarely heterogeneity can co-exist with the presence of various other cellular components or lines of differentiation in MMB, including cells that exhibit neuronal/ganglionic differentiation (9 tumors) or glial/astrocytic differentiation (8 tumors), heterologous elements (3 tumors), and pigmented/ melanotic cells (3 tumors). Likewise, occasional case reports mention discrete islands of rhabdomyoblastic components [40]. Apparantly, there is no cutof level of morphological spectrum. It is worth mentioning that rhabodomyoblasts, melanocytes, and muscle cells are all inherently positive for YAP1 immunostain. Therefore, an indeterminant subtype (YAP1 positive, negative for GAB1, and cytoplasmic beta-catenin) often is observed in cases of MMB [41]. MMB can express different molecular subgroups (e.g., large cell/anaplastic, desmoplastic) [42]. However, the reported outcomes of patients with MMD treated with surgery followed by radiation and/or chemotherapy have been very poor [43-45].
Due to disease rarity, only few reported studies are present. Kathleen et al conducted a retrospective review of the radiographic and pathologic characteristics, treatment, and clinical outcomes of six children with MMB who were treated at St. Jude Children’s Research Hospital (Memphis, TN) between 1984 and 2003 [40]. The median age at diagnosis was 4.5 years Large cell/ anaplastic (five tumors), nodular/desmoplastic (two tumors), and classic (two tumors) medulloblastoma histologies were encountered either alone (five tumors) or in combination with each other (two tumors). All 4 tumors that were tested exhibited alterations in chromosome 17 or c-myc amplification. All patients underwent macroscopic total resection and subsequently received chemotherapy and craniospinal (five patients) or local conformal (one patient) radiotherapy. At a median follow-up of 92 months (range, 23-187 months), 3 patients remain alive with no evidence of disease, 2 patients have died of disease, and 1 patient has died of secondary acute lymphocytic leukemia. None presented with metastasis or relapse during the follow-up. In another large cohort study done by Mahapatra et al. described seven patients (six children and one adult) with MMB. Fourteen who were treated by chemotherapy and CSI for all patients in that series. Of the six patients 14 all died of disease within 3 years of diagnosis. In 1 child, spinal leptomeningeal disease was noted 5 months postdiagnosis. In the remaining patients, however, the pattern of recurrence was not otherwise described [46]. Chowdhury et al. described 3 children with MMB, one patient in that study died of postoperative complications, and the other two patients were alive at the time of the report. Unfortunately, follow-up in that series was quite short, ranging from 1 to 3 months. Non-reported leptomeningeal disease [38]. Compared to our case, the primary cerebellar tumor was of medulloblastoma, desmoplastic/nodular morphology, P53 wildtype, MET point mutation, with very rare myogenic differentiation in the form of strap cells. However, developed relapse in the form of LMD within two years, and the metastatic focus was of pure rhabdomyoblastic differentiation.
This is a unique, un-reported event.
Conclusion
Here we present an unusual presentation of a rare variant of a rare tumor, which is pure rhabdomyoblastic differentation in multiple drop metastasis of a “medullomyoblastoma, desmoplastic/nodular, MET-mutated, P53 wildtype” case within 2 years of diagnosis. This case report is unique as it describes the detailed morphology of the myogenic differetiation types expected to be seen in medulloblastoma transdifferentiation, to enrich the available literature. In addition, the metastatic progression of disease was always assumed to be of the same morphology of the primary, because most likely it is not biopsied, however, this case proves that it can have different morphologies other than the primary, which is even rarer -not even reported- of rhabdomyoblastic nodule. This transdiffrentiation may have an impact on the patient response to further therapy and subsequently disease progression and patient outcome.
Declarations
Ethical approval and consent to participate: Case reports are not applicable for ethical approval by the ethical committee.
Consent for publication/ Conflict of interest: Written informed consent was obtained from the patient for publication of this case report and any accompanying histopathological images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests: No financial and non-financial interests to be declared.
Authors’ contributions: Conceptualization: M. Alm, writing, review and editing: M. Alm. F. dashi, N. Khalifa, R. Mijalcic; supervision: M. Alm. All authors have read and agreed to the published version of the manuscript.
Availability of data and material: The patient’s data and diagnostic material are available and can be provided by hyperlink upon request.
Acknowledgements: Not applicable
Funding: No funds have been received.
References
- Bailey P, Cushing H (1925) Medulloblastoma Cerebelli: A common type of midcerebellar glioma of childhood. Arch Neurol Psychiatry 14(2): 192-224.
- Raffel C (2004) Medulloblastoma: Molecular genetics and animal models. Neoplasia 6(4): 310-322.
- Smoll NR, Drummond KJ (2012) The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J Clin Neurosci 19(11): 1541-1544.
- Smoll NR (2012) Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs). Cancer 118 (5): 1313-1322.
- Franceschi E, Giannini C, Furtner J, Pajtler KW, Asioli S, et al. (2022) Adult Medulloblastoma: Updates on Current Management and Future Perspectives. Cancers (Basel)14(15): 3708.
- Nguyen A, Dada OT, Desai PD, Ricci JC, Godbole NB, et al. (2023) Leptomeningeal Metastasis: A Review of the Pathophysiology, Diagnostic Methodology, and Therapeutic Landscape. Curr Oncol 30(6): 5906-5931.
- Coca-Pelaz A, Bishop JA, Zidar N, Agaimy A, Gebrim EMMS, et al. (2022) Cervical lymphe node metastases from central nervous system tumours: a systematic review. Cancer Manag Res 14:1099-1111.
- Reihanian Zoheir, Hamid Behzadnia, Ahmad Kheiri Namin, Nooshin Zaresharifi (2020) A Rare Case of Medulloblastoma with Supratentorial Metastasis Two Years after Treatment. Arq bras neurocir 39(3): 235238.
- Michalski J, Janss A, Vezina G, et al. (2016) Results of COG ACNS0331: a phase III trial of involved-field radiotherapy (IFRT) and low dose Craniospinal irradiation (LD-CSI) with chemotherapy in average-risk Medulloblastoma: a report from the Children’s oncology group. Int J Radiat Oncol Biol Phys 96(05): 937938.
- Rickert CH (2003) Extraneural metastases of paediatric brain tumours. Acta Neuropathol 105(04): 309-327.
- Young RJ, Khakoo Y, Yhu S, Wolden S, De Braganca KC, et al. (2015) Extraneural metastases of medulloblastoma: desmoplastic variants may have prolonged survival. Pediatr Blood Cancer 62(4): 611-5.
- Garzia L, Kijima N, Morrissy AS, De Antonellis P, Guerreiro-Stucklin A, et al. (2018) A Hematogenous Route for Medulloblastoma Leptomeningeal Metastases. Cell 172(5): 1050-1062.
- Holmberg KO, Borgenvik A, Zhao M, Giraud G, Swartling FJ (2024) Drivers Underlying Metastasis and Relapse in Medulloblastoma and Targeting Strategies. Cancers (Basel) 16(9): 1752.
- Kahn SA, Wang X, Nitta RT, Gholamin S, Theruvath J, et al. (2018) Notch1 regulates the initiation of metastasis and self-renewal of Group 3 medulloblastoma. Nat Commun 9(1): 4121.
- Kahlert UD, Joseph JV, Kruyt FAE (2017) EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol Oncol 11(7): 860-877.
- Huang G, Xu Q, Cui Y, Li N, Bian X, et al. (2016) Medulloblastoma stem cells: Promising targets in medulloblastoma therapy. Cancer Sci 107(5): 583-589.
- Low SYY, Bte Syed Sulaiman N, Tan EEK, Ng LP, Kuick CH, et al. (2020) Cerebrospinal fluid cytokines in metastatic group 3 and 4 medulloblastoma. BMC Cancer 20(1): 554.
- Garzia L, Kijima N, Morrissy AS, De Antonellis P, Guerreiro-Stucklin A, et al. (2018) A Hematogenous Route for Medulloblastoma Leptomeningeal Metastases. Cell 172(5): 1050-1062.
- Zapotocky M, Mata-Mbemba D, Sumerauer, D, Liby P, Lassaletta A, et al. (2018) Differential patterns of metastatic dissemination across medulloblastoma subgroups. J Neurosurg Pediatr 21(2): 145-152.
- Ramaswamy V, Northcott PA, Taylor MD (2011) FISH and chips: The recipe for improved prognostication and outcomes for children with medulloblastoma. Cancer Genet 204(11): 577-588.
- Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, et al. (2013) Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol 14(12):12001207.
- Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, et al. (2016) Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol 131(6): 821-831.
- Cavalli FM, Remke M, Rampasek L, Peacock J, Shih DJ, et al. (2017) Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31(6): 737-754.
- Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, et al. (2013) Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol 14(12):12001207.
- Okonechnikov K, Federico A, Schrimpf D, et al. (2023) Acta Neuropathol Commun 11: 7.
- Marinesco G, Goldstein M (1933) On an anatomical form of medulloblastoma not yet described, medullomyoblastoma. Ann Anat Pathol (Paris) 10: 513-525.
- Mohamed Allaoui, Mustapha Azzakhmam, Mohamed Amine Es-Saoudi, Mohamed Reda El Ochi, Amal Damiri, et al. (2022) Medullomyoblastoma: A Case Report and Literature Review of a very Rare Variant of Medulloblastoma. Sch J App Med Sci 10(4): 510-514.
- Helton KJ, Fouladi M, Boop FA, Perry A, Dalton J, et al. (2004) Medullomyoblastoma: a radiographic and clinicopathologic analysis of six cases and review of the literature. Cancer 101(6): 1445-1454.
- Park SY, Kim JH, Kim KT, Kim YJ, Kim TH, et al. (2004) A case of medullomyoblastoma of cerebellopontine angle mimicking acoustic neuroma. Yonsei Med J 45(4): 719-722.
- Rattenberry W, McDonough CH, Burger PC, Cohen KJ (2011) Medulloblastoma with myogenic differentiation: long-term survival in a patient treated with aggressive combination therapy and autologous stem cell transplantation. J Neurooncol 105(3): 659-662.
- Mahapatra AK, Sinha AK, Sharma MC (1998) Medullomyoblastoma: a rare cerebellar tumour in children. Childs Nerv Syst 14(7): 312-316.
- Ingraham F, Bailey O (1946) Cystic teratomas and teratoid tumours of the central nervous system in infancy and childhood. J Neurosurg 3(6): 511-532.
- Lindberg E, Persson A, Øra I, Mertens F, Englund E, et al. (2007) Concurrent gain of 17q and the MYC oncogene in a medullomyoblastoma. Neuropathology 27(6): 556-560.
- Smith TW, Davidson RI (1984) Medullomyoblastoma: a histologic, immunohistochemical, and ultrastructural study. Cancer 54(2): 323332.
- Stahlberger R, Friede RL (1977) Fine structure of myomedulloblastoma. Acta Neuropathol 37(1): 43-48.
- Walter GF, Brucher JM (1979) Ultrastructural study of medullomyoblastoma. Acta Neuropathol 48(3): 211-214.
- Sachdeva MU, Mahesha Vankalakunti, Aruna Rangan, Bishan D Radotra, Rajesh Chhabra, et al. (2008) The role of immunohistochemistry in medullomyoblastoma-a case series highlighting divergent differentiation. Diagn Pathol l3:18.
- Chowdhury C, Roy S, Mahapatra AK, Bhatia R, et al. (1985) Medullomyoblastoma: a teratoma. Cancer 55(7): 1495-1500.
- Warzok R, Janisch W, Schreiber D (1982) Gliomedullomyoblastoma. A contribution to the morphology and biological behavior of myogenic brain tumors. Zentralbl Allg Pathol 126(1-2): 5-15.
- Helton KJ, Fouladi M, Boop FA, Perry A, Dalton J, et al. (2004) Medullomyoblastoma: a radiographic and clinicopathologic analysis of six cases and review of the literature. Cancer 101(6): 1445-1454.
- Gupta K, Jogunoori S, Satapathy A, Salunke P, Kumar N, Radotra BD, Vasishta RK (2018) Medulloblastoma with myogenic and/or melanotic differentiation does not align immunohistochemically with the genetically defined molecular subgroups. Hum Pathol 75: 26-33.
- Leonard JR, Cai DX, Rivet DJ, Kaufman BA, Park TS, et al. (2001) Large cell/anaplastic medulloblastomas and medullomyoblastomas: clinicopathological and genetic features. J Neurosurg 95(1): 82-88.
- Helton KJ, Fouladi M, Boop FA, Perry A, Dalton J, et al. (2004) Medullomyoblastoma: a radiographic and clinicopathologic analysis of six cases and review of the literature. Cancer 101: 1445-1454.
- Stefanits H, Ebetsberger-Dachs G, Weis S, Haberler C (2014) Medulloblastoma with multi-lineage differentiation including myogenic and melanotic elements: a case report with molecular data. Clin Neuropathol 33(2): 122-127.
- Orr BA (2020) Pathology, diagnostics, and classification of medulloblastoma. Brain Pathol 30(3): 664-678.
- Mahapatra AK, Sinha AK, Sharma MC (1998) Medullomyoblastoma. A rare cerebellar tumour in children. Childs Nerv Syst 14(7): 312-316.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.