Case Report: Activating PIK3CD Mutation in a Patient with Refractory Ascites
by Emadia Elaki1, Abdulwahab Al-Ayoubi1*, Ghannam Al-Ghannam1, Abdulaziz Alsayegh1, Amani Siddig1, Ahmed Alawfi2, Arwa Safar Al Shaibani2, Yasser Al Barrak2, Eid Alrasheedi3, Adel Homoud Almudaibigh4
1Pediatric Allergy &Immunology Department, King Saud Medical City, Riyadh, Saudi Arabia
2Pediatric Gastroenterology King Saud Medical City, Riyadh, Saudi Arabia
3General Pediatric King Saud Medical City, Riyadh, Saudi Arabia
4Pediatric Hematomcology King Saud Medical City, Riyadh, Saudi Arabia
*Corresponding author: Abdulwahab Al-Ayoubi, Pediatric Allergy &Immunology Department, King Saud Medical City, Riyadh, Saudi Arabia.
Received Date: 02 February 2024
Accepted Date: 06 February 2024
Published Date: 08 February 2024
Citation: Elaki E, Al-Ayoubi A, Al-GhannamG, Alsayegh A, et al. (2024) Case Report: Activating PIK3CD Mutation in a Patient with Refractory Ascites. Ann Case Report 9: 1630. https://doi.org/10.29011/2574-7754.101630
Abstract
Background: Activated phosphoinositide 3-kinase delta syndrome (APDS) is a recently described combined immunodeficiency condition resulting from gain-of-function mutations in PIK3CD, which encodes the catalytic subunit of phosphoinositide 3-kinase D (PI3Kd). Mutations in PIK3CD cause primary immunodeficiency with a spectrum of clinical manifestations characterized by recurrent respiratory tract infections, susceptibility to uncontrolled viral infections, impaired vaccine response, autoimmunity, hepatosplenomegaly, and an increased incidence of B-cell lymphoproliferation and lymphoma. Diagnosis of these deficiencies is crucial for their management. In recent years, targeted treatment with selective PI3Kd inhibitors has had a substantial effect on controlling the symptoms of these diseases. Case Presentation: Herein, we report a case of a patient with genetically confirmed PIK3CD mutation who initially presented with generalized lymphadenopathy. Hepatosplenomegaly complicated by immune-mediated anemia and thrombocytopenia resolved after a splenectomy. Over the next few months, the patient developed idiopathic, severe, and refractory chronic ascites requiring repeated diagnostic and therapeutic paracentesis. We hypothesized that autoimmune ascites, after exclusion of common and rare causes of ascites, especially the PIK3CD mutation, is a risk factor for autoimmune complications. The patient responded well to the corticosteroid therapy. Conclusion: PIK3CD mutation is a rare primary immune disease associated with immune dysregulation, immunodeficiency, and malignancy. Autoimmune ascites is an extremely rarely reported complication that needs to be considered after ruling out other known causes.
Keywords: Activated PI3Kδ Syndrome; PIK3CD; Genetic variant; Sirolimus
Introduction
Primary immunodeficiencies are inherited disorders of the immune system that include approximately 500 single-gene inborn errors of immunity [1,2]. Genetic defects in the immune system lead to an increased susceptibility to recurrent, severe, or unusual infections. Recently, germline heterozygous gain-of-function (GOF) mutations in PIK3CD, encoding catalytic p110δ, have been shown to cause a primary immunodeficiency. Mutations in this gene can result in lymphadenopathy and combined immunodeficiency and have been reported to induce immunodeficiency in different ways [3]. Activated phosphoinositide 3-kinase δ syndrome (APDS) is a rare, autosomal dominant primary immunodeficiency caused by GOF mutation in PIK3CD or PIK3R1 genes. Overactivation of this kinase leads to the activation of mTOR, promotion of cell growth and protein synthesis, and inhibition of apoptosis, leading to autoimmunity, lymphoproliferation, and recurrent infections. Paradoxically, both loss-of-function and gain-of-function mutations of these genes lead to immunosuppression through different mechanisms [4,5]. The biochemical and clinical symptoms of patients with APDS1 (PIK3CD mutations) and APDS2 (PIK3R1 mutations) are similar [6,7]. (Tables 1)
|
Laboratory investigations |
During admission and follow-up (before splenectomy) |
After splenectomy |
Recent Lab Values |
Reference |
||
|
White blood cell |
14.34 |
1.84 |
3.74 |
27.78 |
23.9 |
4–12 |
|
Hemoglobin (g/dL) |
7.5 g/L |
7.4 |
9.1 |
11 |
9.98 |
12.9–15.5 |
|
Neutrophils |
5.3 |
0.5 |
3 |
21.2 |
16.6 |
|
|
Platelets |
98 |
29 |
64 |
848 |
596 |
150–350 |
|
Retic count |
7.76 |
5.75 |
7.69 |
|||
|
PT second |
2.3 |
11.6 |
13.3 |
5.54 |
14.2 |
12.3–14.7 |
|
INR |
1 |
1.1 |
1 |
1.1 |
0.8–1.2 |
|
|
PTT second |
38 |
30 |
40 |
34.1 |
26–40 |
|
|
ESR |
120 |
120 |
120 |
120 |
130 |
0–30 mm/h |
|
CRP (mg/L) |
221 |
78 |
9.37 |
75.36 |
10 |
<10 |
|
ALT |
12 |
14 |
4.6 |
2.3 |
43 |
0–41 |
|
AST |
20 |
54 |
13.1 |
18.1 |
42 |
0–40 |
|
ALP |
130 |
107 |
92 |
129 |
170 |
<115 |
|
LDH |
404 |
315 |
100 |
273 |
<248 |
|
|
Amylase |
20 |
11 |
||||
|
Total protein |
(77.3 g/dl) |
|||||
|
Bilirubin total |
(6 µmol\L) |
|||||
|
Immunological work up |
||||||
|
IgA (g/L) |
<0.3 g/dl |
0.21 |
0.34 |
<0.5 |
0.76–3.9 |
|
|
IgG (g/L) |
1.1 g/dl |
2.5 g/dl |
5.3 g/dl |
6.78 g/dl |
7.0–16.0 |
|
|
IgM (g/L) |
41.6 g/dl |
18.81 g /dl |
10 g/dl |
7.34 g/dl |
0.45–2.30 |
|
|
IgE |
6.3 |
|||||
|
CD3 Absolute count |
3517 |
2000–6900 |
||||
|
CD19 Absolute count |
86 |
700–2500 |
||||
|
CD16+CD56 Absolute count |
619 |
100–1000 |
||||
|
CD3+CD4 Absolute count |
581 |
14005100 |
||||
|
CD3+CD8 Absolute count |
2635 |
600–2100 |
||||
|
Sodium |
141 |
139 |
138 |
|||
|
Potassium |
4.6 |
4.8 |
4.87 |
|||
|
Phosphorus |
1.58 |
1.04 |
1.45 |
|||
|
Magnesium |
0.94 |
0.78 |
1.02 |
|||
|
Urea |
1.03 |
3.6 |
1.8 |
|||
|
Creatinine |
18.7 |
18.8 |
24 |
|||
|
Albumin |
29.87 |
31.3 |
32 |
|||
|
Ferritin |
130.2 |
|||||
|
Ammonia |
63.1 |
|||||
|
Anti SSA(RO) |
Normal |
|||||
|
Antibody 1.7 units |
||||||
Table 1: Laboratory Investigation.
Case Presentation
A patient was admitted with septic arthritis when he was 12 months old and received a full course of antibiotics. Two months later, he had a similar episode. At 25 months, he was admitted with severe pneumonia and pleural effusion to the PICU, where he remained for a few days, and then moved to the general ward. At 29 months of age, he was admitted for bronchopneumonia.
At the age of 3 years, he was referred to our hospital, King Saud Medical City, with progressive abdominal distension, hepatosplenomegaly, and cytopenia. He thoroughly examined and common infectious, oncological, and metabolic causes were excluded. The basic immune workup was abnormal and showed high IgM and low IgG and IgA levels. Gene testing confirmed a heterozygous variant of the PIK3CD mutation. Therefore, regular intravenous immunoglobulin, prophylactic trimethoprim-sulfamethoxazole, and a pre-stem cell transplantation workup was initiated. He continued to have recurrent severe anemia and thrombocytopenia that required irradiated PRBC and platelet transfusion and did not respond well to corticosteroid and immunosuppressive therapy. Therefore, the patient underwent a splenectomy, which resulted in a substantial improvement in his blood profile. Rapamycin (sirolimus) was initiated, and the dose was adjusted based on serum drug levels.
However, over the next few months, he developed recurrent severe intractable ascites of undetermined etiology, complicated by peritonitis that required diagnostic and therapeutic ultrasound-guided paracentesis and pigtail drain placement several times (five times a year with approximately 5 L drained each time). In the absence of peritonitis, ascites fluids were transudate, and the serum ascetic albumin gradient (SAAG) was more than 1.1. Extensive workup by different subspecialties was performed, and both common and rare causes of ascites were ruled out. He was evaluated in another tertiary center, and no cause was identified.
Therefore, autoimmunity-related ascites was considered, and oral prednisolone was initiated at 2 mg/kg/d for two weeks then tapered gradually over six months. During the therapy, the patient was ascites-free for six months. However, at the end of corticosteroid tapering (0.125 mg/kg/dose) every other day, the patient experienced ascites relapse. Therefore, the initial prednisolone dose was resumed, and azathioprine was started. The patient was referred to a higher center for stem cell bone marrow transplantation.
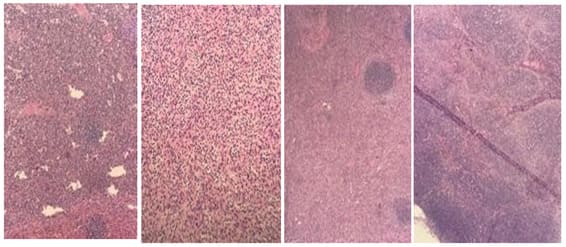
Figure 1: A, B. Part of the spleen; C. Wedge biopsy showing expanded red bulb by sinusoidal dilatation, old hemorrhage (hemosiderin laden macrophages identified), fibrosis (feature of hemolytic anemia), and no malignancy.D. Lymph node biopsy showing reactive lymphoid hyperplasia and no malignancy.
Discussion
We present an overview of the clinical course of a patient with a unique clinical presentation of refractory ascites. Here, we report a case of a heterozygous, pathogenic, variant of PIK3CD, known as Known Saudi Mutation, including deleterious variants of PIK3CD. The present case is worth reporting because of the novelty of one of the clinical presentations of refractory ascites with undetectable causes. To the best of our knowledge, no PIK3CD mutations have been reported in patients with refractory autoimmune ascites.
In 2018, Simone et al. reported a case of 67-year-old male patients with autoimmune ascites who responded well to mycophenolate mofetil. APDS is a heterogeneous inborn error of immunity (IEI), first reported in 2013 [4]. It is caused by autosomal dominant mutations in PIK3CD (resulting in APDS1) or PIK3R1 (resulting in APDS2), which increases the activity of phosphoinositide-3-kinase delta (PI3Kd) [6]. Unfortunately, activating somatic PIK3CD mutations have been associated with variable clinical features, ranging from asymptomatic to severe immunodeficiency, causing profound complications and early death.
The most common clinical manifestation of APDS is recurrent respiratory infection. A complication of frequent respiratory tract infections is bronchiectasis [8], in addition to lymphoproliferative and autoinflammatory disease. Autoimmune presentations are features of APDS1 in up to 42% of cases [9]. Among these, were the most frequent manifestations, followed by Evans syndrome, type 1 diabetes mellitus, enteropathy, arthritis, SLE, autoimmune thyroiditis, sclerosing cholangitis, Sjogren syndrome, and autoimmune hepatitis [10,11].
Lymphoproliferation leads to lymphadenopathy and organomegaly and increases the risk of malignant lymphoma [12,13]. Some patients also experience failure to thrive, neurocognitive development, and syndromic features unrelated to immunodeficiency, especially APDS2, hyperextensibility of short stature, hyperextensibility of joints, and PIK3R1 variants [14].
Whole-genome sequencing is likely to become routine in IEI in the future. The new molecules being approved as targeted therapies will gain importance for the treatment of APDS and IEI in general [15] Genetic testing is a promising approach, and diagnosis via genetic testing is given priority over the clinical and even laboratory phenotypes and has been used successfully in diagnosing rare diseases [16].
Although our patient underwent two lymph node biopsies and a bone marrow biopsy that revealed no malignancies, he still requires long-term follow-up and lymph node biopsies in the future. The risk of malignancy increases with age. His initial presentation was pancytopenia (autoimmune anemia and thrombocytopenia), which responded well to splenectomy.
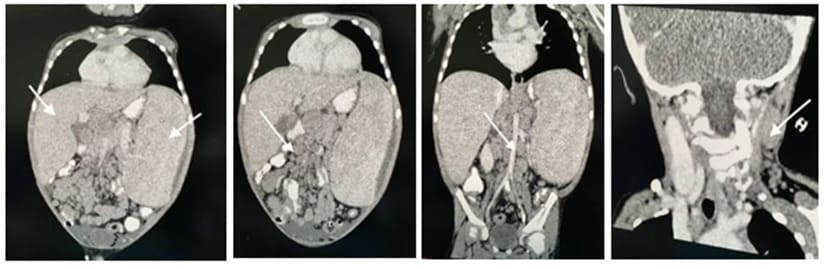
Figure 2: A. This is coronal cut showing a significantly enlarged liver and spleen. B. Abdominal CT showing multiple retroperitoneal lymph nodes. C. Abdominal CT and pelvis showing multiple mesenteric lymph node enlargement.
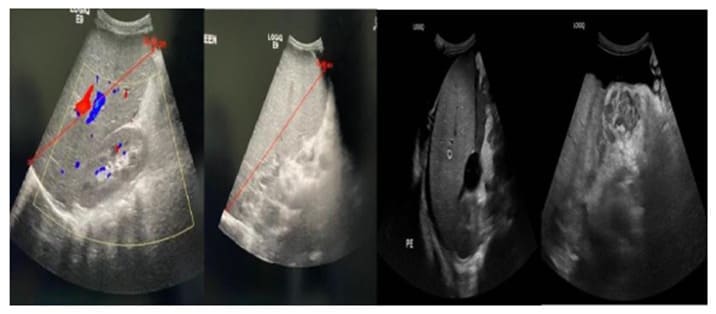
Figure 3: A. Ultrasound of abdomen showing enlarged liver with liver span of 14.03 cm. B. Ultrasound of abdomen showing enlarged spleen 19.45 cm. C. Moderate amount of free ascites fluid noted in the abdomen and pelvis. The largest amount in the left lower quadrant is unchanged compared to previous study.
Patients with APDS display autoimmune phenotypes, with almost 50% of them exhibiting autoimmune-mediated organ damage, such as thrombocytopenia [6]. An overview of this case provides new information regarding the broad spectrum of clinical manifestations of the disease by including ascites as an evolution over time. Rapamycin (sirolimus) was initially administered along with intravenous immunoglobulin and trimethoprim/sulfamethoxazole as prophylaxis during the early stages of genetic testing [17].
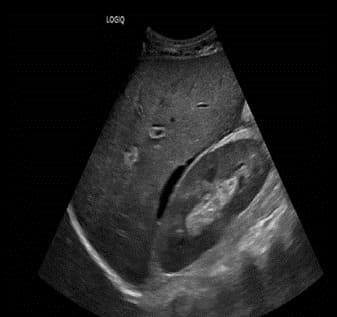
Figure 4: Overall improvement of previous intra-abdominal free ascites as a trace amount of free ascites not amenable for drainage.
The patient was transferred to the KFSH&RC for bone marrow transplantation because hematopoietic stem cell transplantation (HSCT) can be a curative approach. The efficacy and safety of sirolimus remain uncertain in patients with APDS; therefore, long-term follow-up is needed [18]. The recently reported PI3Kδ inhibitor, leniolisib, showed an excellent control of the lymphoproliferation and also improved the cytopenias at the end of treatment [19]. The FDA has approved it as the first-line treatment for activated PI3Kδ syndrome in adult and pediatric patients of 12 years of age [20,21]. Allogeneic HSCT is a curative option; however, it is associated with a mortality rate of approximately 20%, and it is unclear whether severe lung disease can be reversed. Additionally, it can cause graft versus-host disease [22-24].
Reference
- Vanselow S, Wahn V, Schuetz C (2023) Activated PI3Kd syndrome – reviewing challenges in diagnosis and treatment. Front Immunol 14: 1208567.
- Quinn J, Modell V, Orange JS, Modell F (2022) Growth in diagnosis and treatment of primary immunodeficiency within the global Jeffrey Modell Centers Network. Allergy Asthma Clin Immunol 18: 19.
- Zhang Q, Ma H, Ma J, Wang D, Zhao Y, et al. (2019) Clinical and genetic analysis of immunodeficiency-related diseases associated with PIK3CD mutations. Pediatr Invest 2: 257-262.
- Coulter TI, Chandra UM, Bacon CM, Babar J, Curtis J, et al. (2017) Clinical spectrum and features of activated phosphoinositide 3-kinase d syndrome: a large patient cohort study. J Allergy Clin Immunol 139: 597-606.e4.
- Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K (2016) PI3Kδ and primary immunodeficiencies. Nat Rev Immunol 16: 702-714.
- Angulo I, Vadas O, Garcon F, Plagnol V, Leahy TR, et al. (2013) Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science 342: 866-71.
- Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, et al. (2014) Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med 211: 2537- 47.
- Condliffe AM, Chandra A (2018) Respiratory manifestations of the activated phosphoinositide 3- kinase delta syndrome. Front Immunol 9: 338.
- Schworer SA, Francis OL, Johnson SM, Smith BD, Gold SH, et al. (2021) Autoimmune cytopenia as an early and initial presenting manifestation in activated PI3 kinase delta syndrome: case report and review. J Pediatr Hematol Oncol 43: 281-287.
- Bacalao MA, Satterthwaite AB (2020) Recent advances in lupus B cell biology: PI3K, IFNγ, and chromatin. Front Immunol 11: 615673.
- Wang S, Huang Z, Lei Y, Han X, Tian D, et al. (2022) Celastrol alleviates autoimmune hepatitis through the PI3K/AKT signaling pathway based on network pharmacology and experiments. Front Pharmacol 13: 816350.
- Jamee M, Moniri S, Zaki-Dizaji M, Olbrich P, Yazdani R, et al. (2020) Clinical, immunological, and genetic features in patients with activated PI3Kdelta syndrome (APDS): a systematic review. Clin Rev Allergy Immunol 59: 323–33.
- Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, et al. (2016) Clinical and immunologic phenotype associated with activated phosphoinositide 3- kinase d syndrome 2: A cohort study. J Allergy Clin Immunol 138: 210-8.e9.
- Chudasama KK, Winnay J, Johansson S, Claudi T, König R, et al. (2013) SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet 93: 150-7.
- Vanselow S, Hanitsch L, Hauck F, Körholz J, Maccari M-E, et al. (2023) Future directions in the diagnosis and treatment of APDS and IEI: a Survey of German IEI Centers. Front Immunol 14: 1279652.
- Pavey AR, Bodian DL, Vilboux T, Khromykh A, Hauser NS, et al. (2017) Utilization of genomic sequencing for population screening of immunodeficiencies in the newborn. Genet Med 19: 1367-1375.
- Lopez-Nevado M, Gonzalez-Granado LI, Ruiz-Garcıa R, Pleguezuelo D, Cabrera- Marante O, et al. (2021) Allende LM. Primary immune regulatory disorders with an autoimmune lymphoproliferative syndrome-like phenotype: immunologic evaluation, early diagnosis and management. Front Immunol 12: 671755.
- Kangm JM, Kim SK, Kim D, Choi SR, Lim YJ, et al. (2020) Successful sirolimus treatment for korean patients with activated phosphoinositide 3-kinase δ syndrome 1: the first case series in Korea. Yonsei Med J 61: 542-546.
- Rao VK, Webster S, Dalm VASH, Šedivá A, van Hagen PM, et al. (2017) Effective “activated PI3Kdelta syndrome”-targeted therapy with the PI3Kdelta inhibitor leniolisib. Blood 130: 2307-2316.
- Rao VK, Webster S, Sedivá A, Plebani A, Schuetz C, et al. (2023) A Randomized, placebo-controlled, phase 3 trial of PI3Kd Inhibitor leniolisib for activated PI3Kd Syndrome. Blood 141: 971-983.
- Rao K, Webster S, Sediva A, Plebani A, Schuetz C, et al. (2022) Interim analysis of safety and hematological parameters of an ongoing long-Term open-Label extension study of investigational PI3Kd Inhibitor leniolisib for patients with activated PI3K delta syndrome (APDS) through December 2021. Blood 140: 1643-1645.
- Notarangelo LD (2019) Hematopoietic stem cell transplantation for activated phosphoinositide 3- kinase δ syndrome: Who, when, and how? J Allergy Clin Immunol 143: 91-93.
- Al-Mousa H, Al-Saud B (2017) Primary immunodeficiency diseases in highly consanguineous populations from Middle East and North Africa: epidemiology, diagnosis, and care. Front Immunol 8: 678.
- Pavey AR, Bodian DL, Vilboux T, Khromykh A, Hauser NS, et al. (2017) Utilization of genomic sequencing for population screening of immunodeficiencies in the newborn. Genet Med 19: 1367-1375.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.