Autosomal Dominant Polycystic Kidney Disease – Genetics and Cyst Formation
Elizabeth Smith*
Department of Pathology and Anatomy, Eastern Virginia Medical School, USA
*Corresponding author: Elizabeth Smith, Department of Pathology and Anatomy, Eastern Virginia Medical School, USA. Tel: +17574660303; Email: SmithEM@EVMS.EDU
Received Date: 01 December, 2018; Accepted Date: 19 December, 2018; Published
Date: 28 December, 2018
Citation: Smith E (2018) Autosomal Dominant Polycystic Kidney Disease – Genetics and Cyst Formation. Int J Clin Pathol Diagn IJCP-123. DOI: 10.29011/2577-2139.000023
1. Introduction
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is the most common hereditary renal disorder with an incidence rate of 1: 400-1000 individuals. ADPKD causes progressive bilateral renal cysts, kidney enlargement, fibrosis, chronic kidney damage and ultimately progresses to End Stage Renal Disease (ESRD). Cyst formation can occur in extrarenal organs, to include the liver, pancreas and spleen. Other complications of ADPKD include hypertension, cardiac abnormalities (often in response to the hypertension) and brain aneurysms. The average age of disease onset is from 20-40 years old, however there are documented cases of very early onset (in utero to 18 months’ post-partum), and early onset (18 months’ post-partum to 19 years old) [1,2]. Nuances of genetic mutations of PKD1 and PKD2 are involved in phenotypic variation of autosomal dominant polycystic kidney disease. There are multiple postulates as to the specifics regarding cyst formation in polycystic kidney disease, “second-hit”, cAMP and epithelial proliferation. Finally, diagnostic techniques and current treatment options will be discussed. Similar polycystic kidney diseases that will not be thoroughly discussed include Autosomal Recessive Polycystic Kidney Disease (ARPKD), previously known as infantile kidney polycystic disease and non-inherited polycystic kidney disease. ARPKD differs from ADPKD in the genetic mutation of the PKHD1 gene on chromosome 6p21 with a second locus of mutation in the DZIP1L gene, which is unlike ADPKDs mutation of PKD1 gene on PKD1 is located on chromosome 16 (16p13.3) and/or mutation of PKD2 gene on chromosome 4 (4q11) [3]. ARPKD differs genetically, in presentation of disease and in incidence rate (1:20,000). Non-inherited polycystic kidney disease occurs in about 10 percent of patients with ADPKD when a random de novo mutation occurs [4]. Non-inherited polycystic kidney disease has a similar presentation, genetic profile (although not inherited), and the aforementioned incidence rate.
2. Case Study
A 29-year-old man presented to hospital with hypertension. His was experiencing pain and hematuria. Upon imaging, his kidneys were enlarged and showed multiple cystic structures. It was determined he was in End Stage Renal Disease (ESRD). His left kidney was removed. At gross examination, his kidney weighed 1543g, and measured 30 x 15 x 12 cm. A portion of the dictation stated “multiple unilocular cysts filled with clear, pink tinged, straw colored serous fluid. The cystic linings are smooth with no evidence of papillary excrescences. The cystic structures appear to extend throughout the cortex and medulla with scant normal kidney parenchyma identifiable (Figure 1a-c). The cysts appear to extend to, but not involve, the sinus fat.” The patient had not previously been diagnosed with autosomal dominant polycystic kidney disease. However, his presentation was consistent with ADPKD. He is currently on a treatment of dialysis and is awaiting possible transplant.
3. Signs and Symptoms of ADPKD
Polycystic kidney disease manifests its signs and symptoms in multiple ways. The most common symptom is hypertension, occurring in up to 70 percent of patients. Pain and discomfort due to abdominal fullness and distention is the second most common indication with an occurrence rate of 60% [6]. NSAIDs, non-steroidal anti-inflammatory drugs, are not recommended for pain management in ADPKD patients when kidney function is reduced or if needed for chronic pain management [7]. Kidney infections and associated fever and flank pain or “backache” are the third most common set of symptoms and can occur in up to 50 percent patients. Hypertension is caused directly from the development of renal cysts.
As renal cysts develop renin is increased. The renin-angiotensin-aldosterone (RAAS) feedback loop is initiated which increases salt and water retention [9], vascular resistance and cell growth (Figure 2). These factors promote renal cyst proliferation. The second most common symptom is grossly visible hematuria (35-55%), with rupture of the cysts causing pain on presentation. It has been shown that gross hematuria is associated with large >15cm in length kidneys [10]. The presentation of gross hematuria below the age of 30 is linked to worse prognosis. There are mild concentrating effects associated with ADPKD such as increased thirst, polyuria, nocturia, urinary frequency [11]. Renal function remains stable with mild or moderate cyst formation and age. There is tipping point at which the renal function dramatically decreases with age and amount of cyst formation. Nephrolithiasis, renal stone formation, is present in up to 25% of patients. Renal stone composition is 50% uric acid and 50% calcium oxalate, this is unusual in that most renal stones in patients that do not have ADPKD are primarily composed of calcium oxalate. Kidney stones should be suspected in ADPKD patients with acute flank pain. Major extrarenal manifestations that may be related to ADPKD are pericardial effusion, cerebral aneurysms and associated headaches, hepatic and pancreatic cysts, cardiac valve disease, abdominal wall/inguinal hernias and seminal vesicle cysts. These manifestations are evident in up to 40 percent of all ADPKD patients [12].
4. Genetics
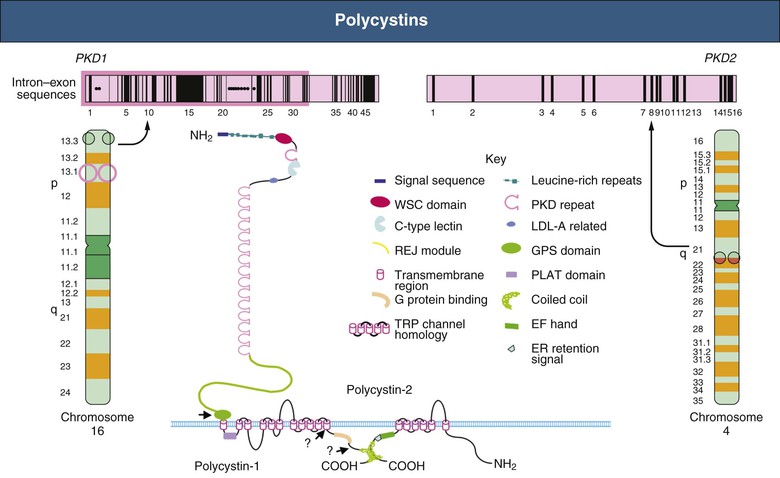
Autosomal dominant polycystic kidney disease is caused by heterogenous mutations of PKD1 and PKD2 (Figure 3). Eighty-five percent of all ADPKD patients have a genetic mutation of PKD1 – ADPKD Type I. PKD1 is located on the short arm of chromosome 16 (16p13.3) and encodes for polycystin-1. PKD1 is a large complex structure that is 46 exons long, of which 1 through 34 exons comprise 2/3 of coding sequence. There are two sub variations in the structure of PKD1, truncated and non-truncated. Truncated PKD1 (frameshift, splicing and nonsense) leads to an earlier onset of End Stage Renal Disease (ESRD) that averages 55.6 years old. The non-truncated PKD1 (missense, in-frame insertion/deletion) leads to the onset of ESRD that averages 67.9 years old [13]. Screening for mutations of PKD1 are difficult due to relative proximity and similarity of nearby genes. The tuberous sclerosis – TSC – (characterized by renal angiomyolipomas and renal cysts) is one such gene that is adjacent to the PKD1 gene. The remaining fifteen percent of ADPKD patients have a genetic mutation of PKD2 – ADPKD Type II. PKD2 is located on the long arm of chromosome 4 (4q11). Mutations in PKD2 lead to ADPKD with a later onset and is less likely to have complications of hypertension, hematuria or progress to end stage renal disease (averaging 79.7 years old). PKD2 is smaller and encodes for polycystin-2. Contiguous deletions of PKD1 and TSC2 and bilineal inheritance of PKD1 and PKD2 increases severity of the disease and is associated with ESRD that occurs in childhood or in utero, versus the average age of ESRD onset of 64 years old [14]. PKD1 missense variants reduce activity and are associated with a milder form of ADPKD, analogous to Type II ADPKD. There is literature that supports that mutations of GANAB, on the long arm of chromosome 11 (11q12.3), in possible conjunction of four additional genes, ALG8, SEC61B, SEC63, and PRKCSH, cause a milder form of ADPKD, Type III and is associated with Autosomal Dominant Polycystic Liver Disease (ADPLD).
PKD1 and PKD2 encode for polycystin 1 and 2 respectively. Polycystins are localized in renal tubular epithelia, hepatic bile ductus and pancreatic ducts. Polycystins are overexpressed in patients with ADPKD due to mutations of PKD1 and PKD2 (Figure 4). Polycystin 1 and 2 interact with endoplasmic reticulum, which is required for maturation of apical and basolateral cell membrane complexes. There is evidence that polycystins are integral membrane proteins in plasma membranes and in primary cilium [16]. Primary cilium is an organelle that arise from the surface of cells and act as a sensor to external signals. These signals are important for correct oriented cell division and in planar polarity. PKD1 molecular production of polycystin 1 has been shown to have cell adhesion properties, which has been attributed to the possible complex of polycystin 1, E-cadherin and catenin. Polycystin 1 has cleavage events at juxtamembrane G-Protein Coupled Receptors (GPCR) proteolytic sites, where no exogenous protease is required, therefore cleavage occurs via “cis-autoproteolysis” [17]. Cleavage at the GPS domain results in 2 fragments that remain non-covalently associated. Thus, abnormalities in the polycystin may reduce cell adhesion properties which may lead to phenotypic changes of ADPKD. PKD2 is involved in Ca signaling, polycystin 2 is expressed in distal tubules, collecting ducts, and thick ascending limb of kidneys. There is strong evidence suggesting polycystin 2 acts as an ion channel. Polycystin-L (homolog to polycystin 2) acts as a channel for calcium, sodium and potassium. PKD2 forms a 6 transmembrane spanning element that is part of a Transient Receptor Protein (TRP) family of non-selective cation channels. Four polycystin 2 channels form a tetrameric structure with voltage sensing domain, pore loop TM5 and TM6 and novel “TOP” domain [11]. This novel “TOP” domain may be important in genetic variation (missense). Polycystin 1 and 2 can form a heterodimer that creates a new, calcium permeable channel.
4.1 Renal Cyst Formation and Pathophysiology
There are over one million nephrons per kidney. There are multiple hypotheses as to the mechanism of cyst formation. Renal cysts are formed in different nephron segments including the collecting tubules. ADPKD is tubular disease with inflammatory components (especially distal tubules and collecting ducts). The idea of marked hyperplasia of epithelia of nephrons and collecting tubules being responsible for cyst formation, is one such hypothesis.
A recent study by Grantham, et al suggests that if a 1mm cyst develops and thus grows to 8cm over 20 years. The cell numbers will increase to 118 million, a 102,000-fold increase [19]. It suggests that early cysts begin as dilations of tubules, constantly being filled by glomerular filtration. Once the cysts grow to a size of 2-3cm, the cysts lose their connection to the functional nephron, and then being filling from secretion of fluid into the cysts. This cyst fluid has been shown (in culture) to harness a lipid secretory ability to promote cyst formation and fluid secretion. (JJ, Evidence for a potent lipid secretagogue in the cyst fluids of patients with autosomal dominant polycystic kidney disease., 1995) [20]. The observation that cysts develop in less than 10 percent of tubules and cystic dilation is focal has led to a “second-hit” theory of cyst formation for ADPKD Type I and Type II. This theory suggests that there is a developmental switch (first hit) and then there is the genetic mutation of PKD1 and PKD2 (second hit). The developmental switch has yet to be determined and may be something as relatively simple as the timing of mutation [21]. There is yet another mechanism of cyst formation that may contribute to ADPKD cyst development. Na-K-ATPase pumps have been identified in ADPKD that are translocated from basolateral membrane to the luminal membrane. This translocation may be attributed to the primary cilia, that has been shown to affect the Planar Cell Polarity (PCP) and subsequently the Oriented Cell Division (OCD (Figure 5)). This process is known as convergent extension [22]. This translocation and conformational change occurs early and promotes sodium and water secretion into the cyst which in turn causes cyst growth.
Another possible mechanism of cyst development is the relationship of cAMP and calcium signaling that induces fluid secretion and cell proliferation. There is a relationship between the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) on the apical membrane of ADPKD cells that acts as cAMP chloride channel.
5. Diagnostic Techniques for ADPKD
Diagnosis of autosomal dominant polycystic kidney disease is reliant on imaging. Ultrasonography is inexpensive, safe and commonly used. Important factors identifying the disease are patient familial history, number and type of cyst and age of presentation. Children, under the age of 18, are not usually screened. However, children that are at risk should be monitored for disease signs and symptoms, i.e. hypertension. Ultrasound imaging criteria for autosomal dominant polycystic kidney disease are based on age and cyst development. Individuals age 18-39 must have at least three unilateral or bilateral kidney cysts, individuals age 40-59 must have at least two cysts in each kidney and individuals aged 60+ must have at least 4 cysts in each kidney. The sensitivity, specificity and positive predictive value is 100 for each age category [19
MRI and CT scanning are more sensitive, but these modalities have been proven to be costly, time-intensive and are without any diagnostic benefits (Figure 6). There are multiple other diseases (localized renal cystic disease, medullary sponge kidney, and multiple benign simple cysts) that can be resemble ADPKD, but careful ultrasonography will exclude these disease processes based on the aforementioned criteria.
Genetic tests and sequencing are performed to diagnosis ADPKD amongst individuals with equivocal imaging results. Sanger sequencing techniques for exon and splice junctions of PKD1 and PKD2 genes is the genetic diagnostic method of choice. PKD1 remains difficult to screen for, due to its size, complexity and proximity to nearby genes. PKD2 is by far the more accommodating gene for genetic screening. Genotype specific disease progression correlated to PKD1 mutations is increased toward ESRD than to PKD2 mutations. Comprehensive mutation screening for ADPKD nonsense, frameshift, spice site alterations are predictive of the truncated/ inactive proteins. Urinary Biomarkers are showing potential for a diagnostic tool [25]. Biomarkers identified in Estimated Glomerular Filtration Rate (eGFR) is a strong predictor but less sensitive in early stages of disease, GFR can remain normal due to hyperfiltration (compensatory function). β2MG (beta 2 micro-globulin) and MCP1 (monocyte chemotactic protein-1) strongly associated with annual change of eGFR (these two biomarkers are associated with inflammation of proximal tubules). Viability of urine biomarkers may not be best choice as frozen storage decreases concentrations for all urinary biomarkers which induces variability. Next generation sequencing is showing promise in enabling high-throughput screening whilst decreasing costs. Targeted next generation experiments are custom designed and read samples based on indexed primers that are then aligned to human reference genome with NCBI database.
6. Treatment and Therapy
There are a variety of treatment options for patients with autosomal dominant polycystic kidney disease which are meant to slow disease progression, however there are no preventative or curative treatments available. Treatments include blood pressure control medication, dietary salt and protein restrictions, and statins. Hypertension can be controlled using Angiotensin-Converting Enzyme (ACE) inhibitors or Angiotensin Receptor Blockers (ARBs) so long as these medications are carefully monitored and there are no contraindications. Use of both ACE inhibitors and ARBs concomitantly has shown no increased advantage [26]. Reduction of salt not only mediates better blood pressure control but has been suggested to moderate progression of kidney disease. Sodium restrictions of ADPKD patients is most often limited to 2 grams per day [27].
Statins are used to aggressively treat hyperlipidemia in patients with ADPKD due to the coronary heart disease equivalence of cystic kidneys. The use of statins have been shown to increase renal flow and endothelial function [28]. There is a dramatic decrease in renal function that corresponds to age, size of kidneys and amount of cyst formation in individuals with autosomal dominant polycystic kidney disease. Fifty percent of all individuals with ADPKD will progress to ESRD needing dialysis, and 10 percent of all dialysis patients have ADPKD. Dialysis is the primary therapy for ADPKD [29]. There are two types of dialysis: peritoneal dialysis and hemodialysis, however peritoneal dialysis in contra-indicated for ADPKD patients with large cysts (decreased peritoneal space) (Figure 7). Hemodialysis is often the therapy of choice. Renal cyst aspiration is performed to treat symptomatic cysts. This is an unrealistic therapy for people with multiple large cysts. Sclerotherapy is used during the procedure to aid in prevention of fluid re-accumulation, and subsequent symptom recurrence [1]. Cyst decortication is a procedure that removes one or more cysts laproscopically and is used primarily for pain relief. This treatment is by far more successful in preventing recurrence than sclerotherapy, 5% vs 82% respectively [30].
Tolvaptan is an approved therapy in the EU and Japan for ADPKD. Clinical studies have shown that Tolvaptan, a vasopressin receptor blocker, slows cyst formation and protects kidney function. Tolvaptan works by blocking vasopressin which blocks cAMP (Figure 8). Tolvaptan is a promising potential treatment for an otherwise hard to treat disease [32].
6.1 Patient Screening and Patient Support
Pre-symptomatic genetic screening tests are not suggested during childhood [12]. However, screening and disclosure of diagnosis of previously undiagnosed children is ultimately the decision of the parents. Screening includes genetic tests for the PKD1 and PKD2 mutations, however due to the limitations of genetic tests up to 15 percent of cases result in false negatives. Post diagnosis “screening” or monitoring for the management of disease progression is suggested in both children and adults and includes blood pressure management, fluid intake monitoring and imaging studies for cyst development and growth. Family planning genetic counselling and PGD/IVF therapy could be options for patients with the PKD1/PKD2 mutations. The decision to use these modalities notwithstanding the availability and cost concerns could show significant medical and societal cost savings. Patient psychological care and support must be provided. ADPKD patients have a high prevalence for anxiety and depression, which must be holistically treated with the disease. Patients should be empowered to advocate for their care and treatment plans and should continue to coordinate resources and relevant expertise [12].
7. Conclusion
Autosomal dominant
polycystic kidney disease has many genetic, cellular and protein complexities
that contribute to the variability and pathogenesis. ADPKD is currently an
incurable condition that progresses to ESRD. The genetics of ADPKD are key to
predicting the onset of disease and novel therapies need to be developed to
slow or halt cyst formation.

Figure 1 A-C: Background has
been modified with Photoshop to remove PHI (AUA, 2018) [
5].

Figure 2: (Bennett, 2009) [8].

Figure 3: (physiopedia, 2018) [15].

Figure 4: (Abdominal Key, 2018) [18].

Figure 5: (HC, 2010) [23].

Figure 6: (Radiopaedia, 2018) [24].

Figure 7:
(Pei, 2018)
[31].

Figure 8:
(Torres VE, 2012)
[33].
4.
NHS
(2016) Autosomal dominant polycystic kidney disease.
5.
AUA
(2018).
{kind=link}
11.
Wilson PD (2004) Polycystic
Kidney disease. N Engl J Med 350: 151-164.
{kind=link}
{kind=link}
{kind=link}
31.
Pei D
(2018) How does the genetic profile of ADPKD impact the disease course.
32.
RK (2013) Tolvapin in ADPKD. Nature Reviews, 1.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.