A Wilms Tumor Patient with Leptomeningeal Metastases, A Case Report and Literature Review of Early CNS Metastasis, At Presentation or During Treatment
by Roos Valk1,2, Francis Wens2*, Annemieke Littooij2,3, Ronald de Krijger2,4, Alida van der Steeg2, Kim Boshuisen2, Sophie van Peer2, Daniëlle Martens5, Nathalie van der Salm6, Jarno Drost2,7, Karin Langenberg2, Lennart Kester2, Geert Janssens2,8, Martine van Grotel2, and Marry van den Heuvel-Eibrink2,9
1University of Utrecht, Utrecht, The Netherlands
2Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands
3Department of Radiology and Nuclear Medicine, University Medical Center Utrecht and Wilhelmina Children’s Hospital, Utrecht, The Netherlands
4Department of Pathology, University Medical Center Utrecht, Utrecht, The Netherlands
5Department of Pediatrics, Isala, Zwolle, the Netherlands
6Department of Pediatrics, University Medical Center Groningen, Groningen, the Netherlands
7Oncode Institute, Utrecht, The Netherlands
8Department of Radiation Oncology, University Medical Center Utrecht, Utrecht, The Netherlands
9Department of Child Health, Wilhelmina Children’s Hospital, University of Utrecht, Utrecht, The Netherlands
*Corresponding author: Francis Wens, Princess Máxima Center for Pediatric Oncology, Heidelberglaan 25, 3584 CS Utrecht, The Netherlands
Received Date: 02 April 2025
Accepted Date: 07 April 2025
Published Date: 09 April 2025
Citation: Valk R, Wens F, Littooij A, de Krijger R, van der Steeg A, et al (2025) A Wilms Tumor Patient with Leptomeningeal Metastases, A Case Report and Literature Review of Early CNS Metastasis, At Presentation or During Treatment. Ann Case Report. 10: 2244. https://doi.org/10.29011/2574-7754.102244
Abstract
Background: Wilms tumor (WT) is the most frequently occurring pediatric kidney cancer. Around 10-17% of patients with Wilms tumor present with metastatic disease, most commonly in the lungs and liver. In patients with Wilms tumor, central nervous system metastases are uncommon. This study was conducted with the aim of identifying patients with leptomeningeal metastases and their specific characteristics.
Methods: An in-depth case description of a patient with a Wilms tumor who developed leptomeningeal metastases during treatment was provided. For this narrative literature review, a PubMed data search was performed up until November 8, 2023. Articles were screened based on predefined inclusion and exclusion criteria.
Results: We report a three-year-old male presenting with stage IV, local stage III intermediate-risk Wilms tumor, with bone metastases and a solitary lung metastasis. Despite the initial response to treatment, the patient developed seizures during therapy, based on leptomeningeal metastases without obvious brain mass. A literature review on early central nervous system metastases in patients with Wilms tumor (at diagnosis, or during therapy) identified 10 additional patients with Wilms tumor showing central nervous system disease but did not identify any other patient with leptomeningeal disease (only).
Conclusions: Leptomeningeal metastases in Wilms tumor patients are rare and may not be recognized due to a lack of available literature and awareness. However, it is crucial to remain vigilant for symptoms of leptomeningeal metastases, even in patients with an intermediate-risk histology.
Keywords: Wilms tumor; leptomeningeal cancer; leptomeningeal metastases; pediatric; Wilms tumor metastases.
Introduction
Wilms tumor (WT), or nephroblastoma, is the most prevalent pediatric kidney cancer. Currently, the overall survival rate for patients with WT is around 90% [1]. Survival depends strongly on biological behavior, associated with histological subtype and staging. Patients with metastatic WT (stage IV) exhibit a 5-year event-free survival rate of about 28-73% [2–5]. Around 10-17% of all patients with WT present with metastatic disease [1,2]. These metastases present most commonly in the lungs and liver and rarely in the bones or in the central nervous system (CNS) [3]. In some patients, CNS metastases are present at diagnosis either as intracerebral metastases or reflect direct spinal infiltration [7,33-36]. Central nervous system metastases can involve the brain parenchyma, leptomeninges (pia, subarachnoid space and arachnoid mater), and the dura.
Here, we describe the characteristics and outcome of a 3-year-old boy that was diagnosed with a stage IV, local stage III intermediate WT who developed diffuse leptomeningeal metastases during treatment. We also pursued and present a narrative review of all patients with WTs with CNS metastases at diagnosis and during treatment, with the aim to identify other patients with leptomeningeal metastases and their specific characteristics, which revealed that leptomeningeal metastases only, had never been reported in children with WT.
Case Presentation
A 3-year-old male, born with a normal birth weight for gestational age, presented with abdominal extension, based on a right-sided renal tumor with a solitary lung metastasis (5 x 4 x 3 mm) and several bone metastases (Figure 1). According to the SIOPRTSG-2016 UMBRELLA protocol (further referred to as UMBRELLA), based on the bone metastases, a tumor biopsy was performed [4]. Histology revealed a WT without anaplasia (Figure 2, Table 1) (Molecular testing showed a MYCN mutation, no 1q gain, and no loss of heterozygosity (LOH) of 16q). The patient started treatment according to UMBRELLA with six weeks of pre-operative chemotherapy, i.e. vincristine, doxorubicin, and actinomycinD (VAD). Repeated imaging showed partial response in week seven (all sites). Tumor-nephrectomy was performed and histology revealed local stage III intermediate-risk histology WT (mixed subtype) (two lymph nodes positive). Post-operative treatment included 34 weeks of chemotherapy (four drugs: carboplatin, etoposide, doxorubicin, and cyclophosphamide), and abdominal radiotherapy at postoperative week 10 (Total of 14.4Gy; lung metastases (CT scan) and bone metastases (total body MRI including brain) showed complete remission at week 10 postoperatively). Nine weeks into post-operative chemotherapy, the patient presented with a mechanical ileus for which adhesiolysis was performed. Radiotherapy to the right flank was given (8 fractions of 1.8Gy). Eighteen weeks into post-operative treatment, the patient experienced afebrile, nocturnal headaches, vomiting, a sudden onset of reduced consciousness (E4M4V1) and seizures, after which he required intubation and PICU admission. MRI of the brain showed diffuse leptomeningeal enhancement with a broad differential diagnosis including infection, inflammation, and metastasis (Figure 1). Antiepileptic drugs, antibiotics, and aciclovir were administered. Upon neurological examination, asymmetry in pupil size (left pupil > right pupil), but no signs of paresis were observed. Reflexes were symmetric. Plantar reflexes were bilaterally indifferent with a tendency towards the Babinski reflex on both sides. Cerebrospinal fluid (CSF) examination revealed an increased protein level (2.23 g/L), glucose <0.6 mmol/L, without elevated pressure measurement. Bacterial cultures, serum galactomannan, and viral PCR were negative. Histology of the CSF showed a few cells, difficult to classify, with some atypia, but did not identify nephroblastoma cells. One day later, post extubation, a cerebral and craniospinal MRI (T2), revealed increased leptomeningeal cerebrospinal enhancement without any CNS mass (Figure 1). Five days later, the patient further developed an unsteady gait and reported dizziness, indicative of progressive ataxia. CSF analysis was repeated, which did not reveal any malignant cells. Since CSF analysis did not provide conclusive results, a leptomeningeal biopsy via thoracal laminotomy was performed, of which histology demonstrated stromal type WT cells (Figure 2). No actionable targets were identified through molecular testing using next-generation sequencing in the personalized medicine program (Figure 3). Fresh, viable material was obtained; however, organoid establishment was unsuccessful, and a compound screen was therefore not feasible [5,6]. Clinically, the headache and vomiting persisted and several atypical multifocal seizures with restless behavior and varying compulsion of the eyes occurred daily. In addition, there was an increase in myoclonus in the right arm and hand. Due to clinically and radiologically confirmed rapid disease progression during therapy, craniospinal radiotherapy was administered and chemotherapy was switched to ifosfamide, carboplatin, and etoposide (ICE), with the aim to administer four courses followed by high-dose chemotherapy (HD-CEM; carboplatin, etoposide, melphalan), and autologous stem cell rescue. After one ICE course, MRI showed a diffuse decrease in leptomeningeal enhancement, intracranially as well as along the spinal axis, and the second ICE course was administered. Unfortunately, after two courses of ICE, craniospinal MRI revealed further progression of leptomeningeal disease. Subsequently, shared decision-making with parents led to the preference for palliative treatment (radiotherapy and oral temozolomide). Fourteen weeks later, a CT scan of the brain showed disease progression with an expansion of ventricles, increased hydrocephalus, and growth of the leptomeningeal metastases. The patient subsequently died due to disease progression (Figure 4).
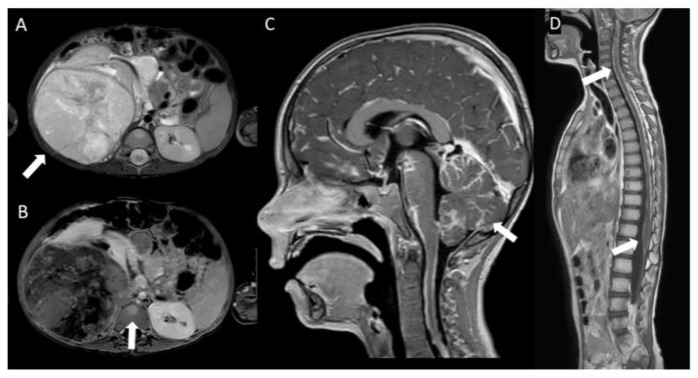
Figure 1: Imaging of Wilms Tumor at primary diagnosis and at leptomeningeal metastases. A: A mass in the left kidney with band-like enhancement, radiologically indicative of a WT. B: Visible on MRI is capsular bleeding, a suspect lymph node, and multiple enhancing lesions in the bone marrow of the pelvis, proximal femora, and spine. C: Abnormally increased leptomeningeal enhancement is observed in the cerebrum. D: Increased enhancement along the spinal cord consistent with the biopsy-proven leptomeningeal disease.
|
Histology |
Microscopic |
TB |
WT without anaplasia (IR-WT) |
|
TN |
Mixed type WT without anaplasia (IR-WT), 2 positive lymph nodes (anterior of right renal artery, and para-aortal) |
||
|
LB |
Histological similarities with the stromal component of the primary WT |
||
|
Immunohistochemistry |
TB |
WT1+, PAX8+, CD56+ blastemal cells, pankeratin+, and EMA+ epithelial cells. INI1 and BRG1 preserved. Myogenin-, MyoD1-, TdT-, CD45-, synaptophysin-, chromogranin-, and S100-. |
|
|
LB |
Desmin+, PAX8+. Focal weak positive for WT1. INI1 and BRG1 preserved. |
||
|
Germline genetics |
No pathogenic variants were detected within the pediatric kidney tumor WES gene panel* |
||
|
Molecular |
TB |
**WES: MYCN variant c.131C>T RNA sequencing: no clinically relevant gene fusions no CNVs, no 1q gain, no LOH 12p, no LOH 16q |
|
|
TN |
**WES: MYCN variant c.131C>T, CTNNB1 variant c.1161T>A RNA sequencing: no clinically relevant gene fusions no CNVs, no 1q gain, no LOH 12p, no LOH 16q, MYCN variant c.131C>T, CTNNB1 variant c.1161T>A |
||
|
LB |
**WES: NRAS variant c.181C>A, gain of chr 2, 8, 13, 14, and 19, no 1q gain, no LOH 12p, no LOH 16q RNA sequencing: NRAS variant c.181C>A |
||
Table 1: Tumor characteristics of current case. Histologic, genetic, and molecular characteristics of the patient described in this report. TB= tumor biopsy, TN= total nephrectomy specimen, LB= leptomeningeal biopsy, IR-WT= intermediate-risk Wilms tumor, WES= whole exome sequencing, CNV= copy number variation, chr= chromosome, LOH= loss of heterozygosity. *Germline gene panel = AMER1, ASXL1, BLM, BRCA2, BUB1B, CDC73, CDKN1C, CEP57, CTR9, DICER1, DIS3L2, FBXW7, GPC3, GPC4, HACE1, MLH1, MSH2, MSH6, NF1, NYNRIN, PALB2, PMS2, PIK3CA, REST, TP53, TRIM28, TRIM37, TRIP13, and WT138. **WES all samples: highly focal CN-LOH on chromosome 11p. Beckwith Wiedeman syndrome negative.
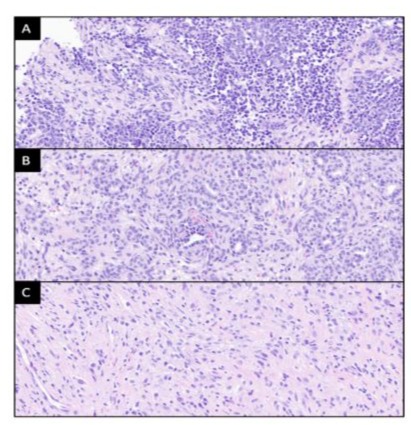
Figure 2: Histology of tumor biopsy, total nephrectomy, and leptomeningeal biopsy. (A) Hematoxylin and eosin-stained images of the initial tumor biopsy showing triphasic Wilms tumor with predominant blastemal component, in addition to minor epithelial and stromal areas. (B) The nephrectomy specimen shows a mixture of mature epithelium and stroma, with only minor areas of blastema (not present in this image). (C) In the leptomeningeal metastases the stromal component predominates, as seen in image. Stromal cells display immunoreactivity for desmin (not shown). All images x400 magnification.

Figure 3: Molecular testing results. Next generation sequencing at molecular testing for both renal tumor (A) and leptomeningeal metastases (B). Renal tumor biopsy molecular testing revealed a MYCN gene mutation, no copy number variations (CNVs), no 1q gain, no LOH 12p, and no LOH 16q. Leptomeningeal biopsy molecular testing revealed an NRAS gene mutation, and a POLE mutation (variant of unknown significance), a gain of chr 2, 8, 13, 14, and 19, no 1q gain, no LOH 12p, and no LOH 16q.

Figure 4: Presentation and evolvement of disease of the patient with WT. Timeline of symptoms, diagnostics, and treatment of the patient described in the current patient case. TB= tumor biopsy, CSF= cerebrospinal fluid, CSF*= cytology: no malignant cells, CSF**= no malignant cells, leptomeninges: stromal component WT, LN+= 2 positive lymph nodes. VAD= cycles of combinations of vincristine + actinomycin + doxorubicin following UMBRELLA protocol. 4Drugs= cycles of combinations of cyclophosphamide + doxorubicin + carboplatin + etoposide following UMBRELLA protocol. ICE= cycles of ifosfamid + carboplatin + etoposide. T= temozolomide, D= dexamethasone 2dd1mg, TN= total nephrectomy, L= laparotomy, RT f = radiotherapy right flank (total cumulative dose 14.4 Gy), RT c + b = radiotherapy craniospinal axis + brain (total cumulative dose 20 Gy).
Narrative literature review
We performed a literature search for the occurrence of CNS metastases at diagnosis or during WT treatment up until November 8, 2023. PubMed database was searched using synonyms for “Wilms tumor”, and “CNS metastases” (full search strategy see Supplementary Table 1). The search yielded 451 articles. All articles, written in English, describing several types of CNS metastases, were eligible for inclusion. Articles concerning nonWilms kidney tumors and all reports describing relapses after the end of therapy, were excluded. After title and abstract screening, a total of 70 articles were eligible for full-text screening, after which 62 articles were excluded. We extracted information on patient characteristics, WT features, treatment, time to CNS metastasis, and the management of CNS metastases, along with their respective outcomes, from the eight included articles. A crossreference check was also performed and yielded two additional articles (Supplementary Figure 1).
Summary of existing cases including literature and our case (n=11)
In the literature, we identified 10 well-documented patients with WT and CNS metastases at diagnosis or during treatment. We summarized clinical characteristics, including the patient described in this report, resulting in 11 patients in total, as depicted in Table 2.
Among all 11 patients, there were six boys and five girls. The median age at WT diagnosis was 4 years (range: 4 months - 14 years), with six out of 11 patients being under five years of age. None of the patients were reported to exhibit an underlying predisposition syndrome associated with WT.
At the time of WT diagnosis, three patients had localized disease, and eight patients had metastasized disease (stage IV) WT (other than CNS, metastatic sites included vertebral bone n=3, bone n=2, pulmonary n=4, liver n=1, and extradural n=1). No patients were reported to have stage I or stage V at the time of diagnosis. Five out of 11 tumors exhibited high-risk histology (blastemal n=3, diffuse anaplastic n=2), and six out of 11 exhibited an intermediate-risk histology. Immunohistochemistry was reported for three patients, revealing WT1 expression in two out of three patients (Table 2). Our patient was the only patient with available molecular testing. He revealed an activating somatic MYCN mutation (c.131C>T (p.Pro44Leu)), no 1qgain, and no LOH of 16q or 1p (Table 1 and 2).
CNS metastases were identified in five patients at WT diagnosis, with the following distribution; epidural (n=1), epidural and intraspinal (n=1), tectal plate and ventricular (n=1), intradural (n=1), and unspecified (n=1). One CNS metastasis caused hydrocephalus and CSF contained tumor cells, however, CNS location was not further specified. Three patients had received preoperative chemotherapy (five did not, and three were not described). After surgery, three patients had received chemotherapy only, and seven had a combination of radiotherapy (brain n=3) and chemotherapy. Therapy for CNS metastasis was not described for the remaining patient.
In six patients (stage II n=1, stage III n=2, and stage IV n=3), CNS metastases (parenchyma n=2, epidural n=3, and in the current patient leptomeningeal n=1) developed during treatment, with a median time from diagnosis to CNS metastasis of 37.5 weeks (3 weeks - 52 weeks). Our patient presented with CNS metastases during treatment for WT, and a combination of metastases also reported by Sudour-Bonnange et al [7]. The latter patient with a blastemal type WT who underwent surgery, received chemotherapy and vertebral radiotherapy, responded well and survived.
All patients exhibited symptoms consistent with CNS involvement, most commonly back pain (n=6) and paresthesia (n=6), at the onset of CNS metastasis (Supplementary Table 2). Epileptic seizures and ataxia were only reported in the current patient and were not documented in the other patients with CNS metastases, at diagnosis or during treatment (Table 3). However, no other patients with WT and leptomeningeal metastases were reported in these studies. Therapy in patients with the development of CNS metastases during treatment encompassed surgery, chemotherapy, or variable combinations of surgery, and/or chemotherapy, and/or radiotherapy, and/or dexamethasone (Table 2).
Five of the 11 patients with WT survived, and six patients died of disease. Of all surviving patients one had favorable histology, one had unfavorable histology with focal anaplasia, and three had blastemal histology subtype. Of all patients who died of disease one had favorable histology, one had mixed histology, one had epithelial histology, two had diffuse anaplastic histology subtypes, and for one patient histology was not described. The patients with blastemal WT survived.
Discussion
We here reported the first patient with WT who developed leptomeningeal metastases during treatment (without CNS mass). At diagnosis, the patient presented with multiple bone metastases and a solitary lung metastasis without signs of CNS metastases. This was in a patient that responded well to treatment (tumor volume decreased from 1236 ml to 560 ml after six weeks of upfront VAD and metastases in the lungs and bones were no longer visible on whole-body MRI and CT thorax), 10 weeks postoperatively.
However, in postoperative week 18, the patient exhibited epileptic seizures and ataxia, due to via laminectomy obtained tissue-proven leptomeningeal disease.
Leptomeningeal metastases in patients with WT have not been previously described in the literature. This absence of prior reports describing leptomeningeal metastases in patients with WT contrasts with their described presence in other pediatric cancers [8,9]. In the current patient, the absence of a tumor mass in adjacent structures, such as bones, makes it unlikely, that the metastasis originated from nearby tissue to the CNS. Despite the protective nature of the blood-brain barrier, which shields the CNS from bloodstream components, metastasis seems to be most likely due to hematogenous spread. Hematogenous spread is described via arachnoid vessels or choroid plexus and also speculated via the retrograde venous pathways along the Valveless Batson’s venous plexus [10–12]. Considering the complexity of breaching the blood-brain barrier, spreading to the CNS without a primary tumor nearby points to the adaptability of tumor cells [13].
WTs are known to metastasize predominantly to the lungs and liver [4]. Bone metastases are rare, having been reported in only 0.44% of WT cases, with a higher likelihood of occurrence at relapse, namely in 76% of bone metastatic cases [14,15]. Additionally, intracranial metastases in WT are very uncommon, occurring in less than 1% of patients with WT, and if so they mainly appear at relapse [3,16,17]. Interestingly, our patient presented with multiple bone metastases at diagnosis. In our case, we had not searched for CNS metastases, and after 18 weeks into postoperative therapy, seizures appeared, for which enhanced MRI was necessary to identify the metastases. It needs to be determined whether, in patients with bone metastasis, baseline staging with this radiological tool should include the CNS screening as standard of care, as bone metastasis has been described to occur together with CNS involvement in several cases now. However, it is unclear whether our patient had the metastases at presentation, and whether we would have been able to rescue the (otherwise well-responding) patient by application of CNS irradiation at an earlier stage. The disease progression of our patient case illustrates the aggressiveness of the disease, despite the initial sensitivity and response to standard treatment with chemotherapy for WT.
Leptomeningeal metastases typically arise as a late-stage complication of cancer, leading to increased intracranial pressure and neurological symptoms [12]. Cancer cells infiltrate the leptomeninges and CSF via direct invasion from adjacent structures, hematogenous spread, or entry through the choroid plexus [10]. Metastatic leptomeningeal cancer cells exhibit distinct phenotypic characteristics and can be localized in the CSF or adhere to the pia and arachnoid mater, respectively ‘floating cells’ or ‘pia-attached’ (MRI positive) cancer cells. Patients with CSF, i.e. floating cell, leptomeningeal metastases have a worse prognosis [18]. Overall, leptomeningeal metastases result in poor overall survival in other cancers, with survival periods ranging from 2.6 to 6.9 months [19].
Our patient developed clinical symptoms based on leptomeningeal metastases without apparent CNS mass. Our current literature review on CNS positive WTs, shows that only 11 well-documented de novo patients (including our patient) have been reported so far. The ten published cases presented with tumor mass. High-risk (HR) tumor characteristics, such as advanced stage, blastemal or diffuse anaplastic histology subtype associated with a more aggressive course, are prevalent. Again, the occurrence of bone metastases seems to be more likely in WT patients with CNS metastases (three out of 11 patients), compared to the described 0.44% in sporadic patients with WT [14]. Recurrent molecular characteristics, such as loss of p53 function in anaplastic tumors, 1p and 16q loss, and 1q gain, potential indicators of poor outcomes, were not studied and thus reported in any, apart from our, CNS metastatic WTpatient [20–23]. No pathogenic germline variants were detected in our patient (Table 1). In our patient, a MYCN somatic mutation was identified in the WT, however not in the leptomeningeal biopsy, where a de novo NRAS somatic mutation was detected. MYCN mutations in WTs are associated with relapses and poor outcome [22,24]. Research into the biological characteristics and development of kidney tumors (SIOP) and relapses is expanding, and research performed by the COG (Children’s Oncology Group) provides indications of an association between specific gene expression signatures and relapses in stage III favorable histology (FH)-WT [25]. However, due to the rarity, studies on the molecular characteristics, that play a role in the development of homing of WT cells towards the CNS specifically, are not to find [2,25]. Hence, further studies are needed to identify molecular and tumorassociated indicators for CNS metastases.
Our patient presented with epileptic seizures and ataxia leading to a suspicion of CNS involvement. Nevertheless, brain CT as well as CSF examination did not explain the patient’s symptoms. CSF cytology is the gold standard for the identification of leptomeningeal metastases, however, it only has a sensitivity of 44-67% for detecting leptomeningeal metastases [26]. CSF diagnostics can detect the freely floating cancer cells, however, the pia mater attached cancer cell form of leptomeningeal metastases is better visualized on MRI [18]. Therefore, even though leptomeningeal metastases are very rare and have not previously been described in patients with WT, given the severity, it is important to perform further diagnostics starting with contrast-enhanced MRI. If a combination of MRI and CSF analysis is inconclusive, our patient case shows, that leptomeningeal biopsy to detect leptomeningeal metastases may be of help.
Leptomeningeal metastases are challenging to treat, and the outcome of CNS disseminated disease seems poor. Early identification and CNS penetrating treatment focuses on postponing neurological deterioration and is important, as performance status expressed in Lansky score at diagnosis is described to be the most important prognostic factor [27,28]. For the treatment of CNS metastases, it is crucial that CNS-penetrating chemotherapeutic agents are administered. This is guaranteed in the current SIOP RTSG Umbrella protocol, partly in upfront treatment through vincristine, but even better in the 4-drug regimen that includes etoposide and carboplatin [4]. However, crossing the blood-brain barrier is challenging and several compounds have a difference in distribution, resulting in different CSF plasma ratios, which affects the treatment efficiency [29]. Regarding the performance status, our patient received upfront therapy with VAD based on WT stage IV with lung and bone metastases. For WT patients with bone metastases, it is currently under investigation, whether the poor outcome that, as reported by the COG renal tumor committee (survival rate only 0-13%) represents a WT subset survival rate of 0-13%, also represents a subset of such poor children in the SIOP_RTSG where pre-operative chemotherapy is applied, and whether a more intensive chemotherapy approach should be considered [4,30,31]. Whether the co-presence of CNS disease is an independent adverse prognostic factor, will be obviously difficult to explore, given the rarity of these cases.
Hence, our study is limited by the scarcity of available studies on patients with WT and CNS metastases, and in particular leptomeningeal metastases. Also, most of these studies did not describe molecular and tumor characteristics. Therefore, identifying indicators for CNS metastases in patients with WT is not conclusive using these limited studies. More, at least, descriptive studies and in-depth molecular and tumor characteristics of CNS metastasized WT patients are needed for early identification of CNS metastases in WTs. Although growing epithelial, stromal, and blastemal components in WT organoids is promising, the organoids from this patient did not grow sufficiently, and compound screening for personalized medicine was not feasible. However, for future exploration of individual treatment options, use of 3D patient derived models will remain of value [32].
|
Reference |
Gender |
Age at Dx |
Synd |
Stage (non CNS sites) |
Histology before treatment |
Molecular (+/-) |
Pre-op |
Surgery |
Postop |
Time to CNS metastases |
CNS location of metastases |
Treatment CNS metastases |
Other metastases at CNS metastasis |
Outcome |
|
[7] |
F |
5.5y |
ND |
IV (B/P/V) |
BT* |
ND |
VAD |
TN |
CT, RT |
Dx |
epidural |
S, CT, RT |
B/P/V |
A |
|
[33] |
F |
14y |
No |
IV (extradural/PaLN/V) |
BT, FH** |
ND |
ND |
TN |
RT, VDD > AADD |
Dx |
epidural, intraspinal |
S, RT, VDD > AADD |
extradural/ PaLN/V |
A |
|
[34] |
M |
8m |
ND |
IV (-) |
NC |
ND |
No |
TN |
VAdC |
Dx |
tectal plate, ventricle |
S, VAdC, RT |
No |
DOD |
|
[35] |
M |
5.5y |
ND |
IV (ND) |
DA |
ND |
ND |
ND |
ND |
Dx |
intradural |
Dm, RT |
ND |
DOD |
|
[36] |
M |
4m |
ND |
IV (extrarenal extension) |
DA |
ND |
No |
TN |
AV |
Dx |
unspecified (CSF) |
ND |
ND |
DOD |
|
[37] |
F |
18m |
ND |
III |
FH |
ND |
ND |
TN |
VAD, ICE, RT |
39w |
Brain (frontal region) |
S, ICE |
No (history of P) |
A |
|
[38] |
M |
20m |
ND |
III |
FH |
ND |
VCD >VDa |
Yes, ND |
Da |
+/- 52w |
cerebellar |
S, RT |
No |
DOD |
|
[39] |
M |
8y |
ND |
IV (P/L/ PaLN) |
ET |
ND |
No |
TN |
AVAC, RT |
3w |
epidural |
RT, JET |
V, L, PaLN |
DOD |
|
[40] |
F |
5y |
ND |
IV (P) |
BT |
ND |
No |
TN |
RT, VAD |
36w |
epidural |
S, RT, VEIE |
No |
A |
|
[41] |
F |
4y |
ND |
II |
UH, FA |
ND |
No |
TN |
RT, DVD |
39w |
epidural |
Dm, S, RT, CT (>CVD) |
Extradural/P |
A |
|
Cur- rent case |
M |
3y |
No |
IV (B/V/P) |
mixed*** |
MYCN muta- tion, no CNVs/1q gain/ LOH 12p/LOH 16q |
VAD |
TN |
RT, 4D |
26w |
leptomeningeal |
ICE, RT |
No |
DOD |
|
* immunohistochemistry: strong WT1+ expression, ** immunohistochemistry: cytokeratin+, synaptophysin-, neurofilament protein-, CD99-, *** Mixed, sub-classification stromal Wilms tumor (score 0.77). Immunohistochemistry: WT1+, PAX8+, CD56+, pankeratin+, EMA+. INI1/BRG1 preserved. Myogenin-, MyoD1-, CD45-, synapto-, chromo-, s1000 Synd= syndrome, CNS= central nervous system, pre-op= pre-operative treatment, post-op= post-operative treatment, M= male, F= female, ND= not described, BT= blastemal, ET= epithelial, ST= stromal FA= focal anaplasia, DA= diffuse anaplastic, FH= favorable histology, UH= unfavorable histology, LOH= loss of heterozygosity, NC= not classified, RT= radiotherapy, CT= chemotherapy, S= surgery, TN= total nephrectomy, Dx= at diagnosis, A= alive; DOD= died of disease, DM= dexamethasone, AV= actinomycin and vincristine; VAD= vincristine, actinomycin-D, and doxorubicin, AVAC = actinomycin, vincristine, doxorubicin, and cyclofosfamide, Da = dactinomycin, 4D = carboplatin, etoposide, doxorubicin, and cyclophosphamide, ICE = ifosfamide, carboplatin, and etoposide, VCD= vincristine, cyclophosphamide, and doxorubicin; VDa= vincristine and dactinomycin, JET = high-dose carboplatin and etoposide, VEIE= vincristine, etoposide, ifosfamide, and epirubicin, DVD= dactinomycin, vincristine sulfate, and doxorubicin hydrochloride, CVD=cyclophosphamide, vincristine sulfate, and doxorubicin hydrochloride, VAdC = vincristine, Adriamycin, and cyclophosphamide, VDD = vincristine, dactomycin, and doxorubicin, AADD= actinomycin-D, adriamycin, dactomycin, and doxorubicin, V= vertebral; P= pulmonal, B= bone other than vertebral metastases, L= liver, PaLN= para-aortic lymph nodes, >= switch of chemotherapy |
||||||||||||||
Table 2: Characteristics and outcome of WT with CNS metastases at diagnosis (Dx) or progression under therapy. Clinical patients and tumor characteristics of all well-documented CNS metastases in patients with WT diagnosed with or presenting during treatment for WT.
|
Search strategy for Pubmed database |
|
1. Wilms tumor: (wilms tumor) OR (wilms’ tumor) OR (wilm tumor) OR (wilm’s tumor) OR (nephroblastoma) |
|
2. Metastases: (metastases) OR (metastasis) OR (carcinomatosis) |
|
3. Central nervous system: (leptomeningeal) OR (meningeal) OR (brain) OR (intracranial) OR (cerebral) OR (spine) OR (spinal) OR (spinal column) OR (vertebral column) OR (vertebra) OR (vertebrae) OR (bone) OR (epidural) OR (intradural) OR (extradural) |
Supplementary Table 1: Search strategy. Search strategy for Pubmed database with a combination search: 1 AND 2 AND 3.

Supplementary Figure 1: Flowchart searching and selection strategy. The initial Pubmed article search identified 451 articles. Title, abstract, and full-text screening on inclusion and exclusion criteria yielded 8 articles, on which cross-reference checking was applied. In total 10 articles were included in this narrative review of literature.
|
Reference |
Time of presentation of CNS involvement |
Back pain |
Leg pain |
Weak- ness of lower limbs |
Uro- genital symptoms1 |
Signs of intracranial pressure2 |
Diminished/ absent tendon reflexes |
Paresthesia / paresis / paralysis3 |
Epi- leptic seizures |
Other4 |
|
|
At Dx |
during treatment |
||||||||||
|
[7] |
x |
x |
x |
x |
|||||||
|
[33] |
x |
x |
x |
||||||||
|
[34] |
x |
x |
x |
||||||||
|
[35] |
x |
x |
x |
x |
x |
x |
|||||
|
[36] |
x |
x |
x |
||||||||
|
[37] |
x |
x |
|||||||||
|
[38] |
x |
x |
x |
x |
|||||||
|
[39] |
x |
x |
x |
||||||||
|
[40] |
x |
x |
x |
x |
x |
||||||
|
[41] |
x |
x |
x |
x |
|||||||
|
current patient |
x |
x* |
x |
x* |
|||||||
|
All symptoms reported just before or at the moment of diagnosis of central nervous system metastasis in patients with Wilms tumor. 1 sphincter disorder [7], urinary incontinence [40,41] 2 headache*, nausea/emesis [38]*, vomiting*, sunset phenomenon [34], hydrocephalus [36,38]* 3 paraparesis causing loss of ambulation [7], unable to walk [38,41], numbness in feet [35], lower limb paralysis [35], complete flaccid paraplegia [35], sensory loss/neuropathy [35], neuropathy [38], spastic paraplegia [41] 4 tiredness [33], drowsiness [38], unsteadiness [38], myoclonus/ataxia*, greater head circumference/size [34,36], muscle spasm [35], inappropriate ADH secretion [36] CNS= central nervous system, Dx= at diagnosis |
|||||||||||
Supplementary Table 2: Presenting symptoms CNS metastases in WT at diagnosis or progression under therapy.
Conclusions
We report the first patient with stage IV, local stage III IR WT, who developed leptomeningeal metastases during treatment. Our literature review indicates that, in general, any CNS metastasis in patients with WT at diagnosis or during therapy is rare, are shown by a mass on imaging, and that leptomeningeal metastases in patients with WT may not be recognized due to the fact that contrast enhanced MRI is not part of our regular diagnostic workup, especially in non-symptomatic disease, and given the rarity of this metastatic site. Because of the reported adverse outcomes in WT patients with CNS metastases, it is crucial to remain vigilant for symptoms that may reflect CNS metastases, especially in children with bone metastasis, even in patients with intermediaterisk histology.
Author contributions: Conceptualization, Martine van Grotel and Marry van den Heuvel-Eibrink.; Methodology, Roos Valk, Francis Wens, Martine van Grotel, and Marry van den Heuvel-Eibrink; Software, not applicable; Validation, in cases of uncertainty regarding article inclusion, Martine van Grotel and Marry van den Heuvel-Eibrink were consulted for independent advice for validation of formal analysis.; Formal Analysis, Roos Valk and Francis Wens.; Investigation, Annemieke Littooij, Ronald de Krijger, Alida van der Steeg, Kim Boshuisen, Sophie van Peer, Jarno Drost, Karin Langenberg, Daniëlle Martens, Nathalie van der Salm, Lennart Kester, Geert Janssens, and Martine van Grotel.; Resources, all authors.; Data Curation, all authors contributed to data collection regarding the reported patient. The data extraction was performed by Roos Valk and Francis Wens for the narrative review.; Writing – Original Draft Preparation, Roos Valk, Francis Wens, Martine van Grotel, and Marry van den Heuvel-Eibrink.; Writing – Review & Editing, all authors.; Visualization, Roos Valk, Francis Wens, Annemieke Littooij, Ronald de Krijger, Sophie van Peer, and Karin Langenberg.; Supervision, Martine van Grotel and Marry van den Heuvel-Eibrink.; Project Administration, Marry van den Heuvel-Eibrink.; Funding Acquisition, not applicable.
Funding: This research received no external funding.
Data availability Statement: These data were derived from resources available in the public domain [see references].
Conflict of Interest statement: The authors have no conflicts of interest to disclose.
References
- Verschuur A, Tinteren HV, Graf N, Bergeron C, Sandstedt B, et al (2012) Treatment of Pulmonary Metastases in Children with Stage IV Nephroblastoma With Risk-Based Use of Pulmonary Radiotherapy. JCO. 30:3533-3539.
- Brok J, Treger TD, Gooskens SL, van den Heuvel-Eibrink MM, Pritchard-Jones K. (2016) Biology and treatment of renal tumours in childhood. European Journal of Cancer. 68:179-195.
- van den Heuvel-Eibrink M m., Graf N, Pein F, Sandsstedt B, Tinteren HV, et al. (2004) Intracranial relapse in Wilms tumor patients. Pediatric Blood & Cancer. 43:737-741.
- Heuvel-Eibrink MM van den, Hol JA, Pritchard-Jones K, Tinteren HV, Furtwangler R, et al. (2017) Position paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nature Reviews Urology. 14:743-753.
- Calandrini C, Schutgens F, Oka R, Candelli T, Mathijsen L, et al. (2020) An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat Commun.11:1310.
- Calandrini C, Drost J. (2022) Normal and tumor-derived organoids as a drug screening platform for tumor-specific drug vulnerabilities. STAR Protoc. 3:101079.
- Sudour Bonange H, Leblond P, Cellier C, Vinchon M, Lervet C, et al. (2015) Bone Vertebrae Metastases With Spinal Cord Compression: A Rare Event in Wilms Tumor. Journal of Pediatric Hematology/ Oncology. 37: e387-e389.
- Chamberlain MC. (1995) Topical Review: A Review of Leptomeningeal Metastases in Pediatrics. J Child Neurol. 10:191-199.
- Cocito C, Martin B, Giantini-Larsen AM, Aspergren MV, Souweidane MM, et al. (2023) Leptomeningeal dissemination in pediatric brain tumors. Neoplasia.;39:100898.
- Nguyen A, Nguyen A, Dada OT, Desai PD, Ricci JC, et al. (2023) Leptomeningeal Metastasis: A Review of the Pathophysiology, Diagnostic Methodology, and Therapeutic Landscape. Current Oncology. 30:5906-5931.
- Le Rhun E, Taillibert S, Chamberlain MC. (2013) Carcinomatous meningitis: Leptomeningeal metastases in solid tumors. Surg Neurol Int. 4S265-S288.
- Grossman SA, Krabak MJ. (1999) Leptomeningeal carcinomatosis. Cancer Treatment Reviews. 25:103-119.
- Wrobel JK, Toborek M. (2016) Blood-brain Barrier Remodeling during Brain Metastasis Formation. Mol Med. 22:32-40.
- Lawler W, Marsden HB. (1979) Bone metastases in children presenting with renal tumours. J Clin Pathol. 32:608-615.
- Iaboni DSM, Chi Y, Kim Y, Dome JS, Fernandez CV. (2019) Outcome of Wilms tumor patients with bone metastasis enrolled on National Wilms Tumor Studies 1‐5: A report from the Children’s Oncology Group. Pediatric Blood & Cancer. 66: e27430.
- Venkatramani R, Chi YY, Coppes MJ, et al. (2017) Outcome of patients with intracranial relapse enrolled on national Wilms Tumor Study Group clinical trials: Venkatramani et al. Pediatr Blood Cancer. 64:e26406.
- Borni M, Kammoun B, Kolsi F, Abdelhedi A, Boudawara MZ. (2018) Isolated central nervous system metastasis in pediatric Wilms’ tumor: A case report and review of literature. Urol Case Rep. 21:78-80.
- Remsik J, Chi Y, Tong X, Sener U, U, Derderian C, et al. (2022) Leptomeningeal metastatic cells adopt two phenotypic states. Cancer Reports. 5: e1236.
- Nayar G, Ejikeme T, Chongsathidkiet P, Elsamadicy A, Blackwell K, et al. (2017) Leptomeningeal disease: current diagnostic and therapeutic strategies. Oncotarget. 8:73312-73328.
- Vujanić GM, Sandstedt B, Harms D, et al. (2002) Revised International Society of Paediatric Oncology (SIOP) working classification of renal tumors of childhood. Medical and Pediatric Oncology. 38:79-82.
- Vujanic GM, Gessler M, Ooms AHAG, et al. (2018) The UMBRELLA SIOP-RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nature Reviews Urology. 15:693-693.
- Williams RD, Chagtai T, Alcaide-German M, et al. (2015) Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget. 6:7232-7243.
- Zheng H, Liu J, Pan X, Cui X. (2023) Biomarkers for patients with Wilms tumor: a review. Frontiers in Oncology.
- Gadd S, Huff V, Skol AD, et al. (2022) Genetic changes associated with relapse in favorable histology Wilms tumor: A Children’s Oncology Group AREN03B2 study. Cell Rep Med. 3:100644.
- Huang CC, Gadd S, Breslow N, Cutcliffe C, Sredni ST, et al. (2009) Predicting Relapse in Favorable Histology Wilms Tumor Using Gene Expression Analysis: A Report from the Renal Tumor Committee of the Children’s Oncology Group. Clin Cancer Res. 15:1770-1778.
- Pellerino A, Brastianos PK, Rudà R, Soffietti R. (2021) Leptomeningeal Metastases from Solid Tumors: Recent Advances in Diagnosis and Molecular Approaches. Cancers. 13:2888.
- Le Rhun E, Preusser M, van den Bent M, Andratschke N, Weller M. (2019) How we treat patients with leptomeningeal metastases. ESMO Open. 4: e000507.
- Brower JV, Saha S, Rosenberg SA, Hullett CR, Ian Robins H. (2016) Management of leptomeningeal metastases: Prognostic factors and associated outcomes. Journal of Clinical Neuroscience. 27:130-137.
- Triarico S, Maurizi P, Mastrangelo S, Attinà G, Capozza MA, Ruggiero A. (2019) Improving the Brain Delivery of Chemotherapeutic Drugs in Childhood Brain Tumors. Cancers. 11:824.
- Bond JV, Martin EC. (1975) Bone metastases in Wilms’ tumour. Clin Radiol. 26:103-106.
- Marsden HB, Lennox EL, Lawler W, Kinnier-Wilson LM. (1980) Bone metastases in childhood renal tumours. Br J Cancer. 41: 875-879.
- Brok J, Mavinkurve-Groothuis AMC, Drost J, Perotti D, Geller JI, et al. (2021) Unmet needs for relapsed or refractory Wilms tumour: Mapping the molecular features, exploring organoids and designing early phase trials – A collaborative SIOP-RTSG, COG and ITCC session at the first SIOPE meeting. European Journal of Cancer. 144:113-122.
- Watanabe R, Takahashi A, Suzuki M, et al. (2023) Adolescent Wilms Tumor With Intraspinal and Bone Metastases: A Case Report and the Review of Literature.
- Harada K, Nishizaki T, Kwak T, Fujisawa H, Nishikawa M, Ito H. (1999) Intracranial Metastasis of Wilms’ Tumor Involving the Tectal Plate without Pulmonary Involvement: Case Report. Pediatric Neurosurgery. 30:331-334.
- Ebb DH, Kerasidis H, Vezina G, Packer RJ, Carabell S, Ivy P. (1992) Spinal cord compression in widely metastatic Wilms’ tumor. Paraplegia in two children with anaplastic Wilms’ tumor. Cancer. 69:2726-2730.
- Kalousek DK, de Chadarévian JP, Mackie GG, Bolande RP. (1977) Metastatic infantile Wilms’ tumor and hydrocephalus; a case report with review of the literature. Cancer. 39:1312-1316.
- Arjmandi K, Rostami T, Yusefian S, Miri-Aliabad G, Kiumarsi A, et al (2015) Brain Metastasis in Wilms’ Tumor: a Case Report. Acta Med Iran. 53:731-732.
- Lowis SP, Foot A, Gerrard MP, Imeson J, Middleton H, et al. (1988) Central nervous system metastasis in Wilms’ tumor: a review of three consecutive United Kingdom trials. Cancer. 83:2023-2029.
- Kurosawa H, Kurumada H, Haga E, et al. (1996) Epidural metastasis in chemoresistant Wilms’ tumor with perilobar nephroblastomatosis. Pediatr Surg Int. 11:153-155.
- Darendeliler E, Ayan I, Kebudi R, Ağaoğlu L, Bilge N, Kinay M. (1994) Epidural metastasis in Wilms’ tumor: a case report and review of the literature. Med Pediatr Oncol. 23:60-63.
- Bever CT, Koenigsberger MR, Antunes JL, Wolff JA. (1981) Epidural metastasis by Wilms’ tumor. Am J Dis Child. 135:644-646.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.