A Rare Case of Abetalipoproteinemia in a Young Sudanese Male: Clinical Challenges and Management
by Ibrahim Babiker Mohamed Seddig1*, Mustafa Mohamed Ali Hassan1, Israa Bader Eldin Ahmed NorEldin1, Motwakil Imam Awadalkareim2
1Internship doctors, El Mek Nimir University Hospital, Sudan
2Associate Professor of Internal Medicine, Faculty of Medicine, Shendi University, Consultant physician, Elmek Nimer University Hospital, Sudan
Motwakil Imam Awadalkareim-ORCID ID: 0000-0002-46799255
*Corresponding author: Ibrahim Babiker Mohamed Seddig, Internship doctors, El Mek Nimir University Hospital, Sudan
Received Date: 14 March 2025
Accepted Date: 18 March 2025
Published Date: 27 March 2025
Citation: Seddig IBM, Hassan MMA, NorEldin IBEA, Awadalkareim MI (2025) A Rare Case of Abetalipoproteinemia in a Young Sudanese Male: Clinical Challenges and Management. Arch Surg Clin Case Rep 8: 244. https://doi.org/10.29011/2689-0526.100244
Abstract
Background: Abetalipoproteinemia (ABL) is an uncommon" autosomal recessive genetic disorder that hinders the absorption of dietary lipids and fat-soluble vitamins, leading to various systemic symptoms. The disorder primarily arises from mutations in the microsomal triglyceride transfer protein (MTTP) gene, leading to a deficiency in apolipoprotein B-containing lipoproteins. This case report outlines the clinical features, diagnostic approach, and therapeutic management of a 33-year-old "Sudanese male with ABL.
Case Presentation: The patient, born to consanguineous parents, exhibited progressive lower extremity edema, facial swelling, nyctalopia, and impaired motor coordination. He had a history of recurrent steatorrhea since early childhood. Clinical evaluation revealed pallor of the palms, koilonychia, onycholysis, fine, brittle hair, and ophthalmoplegia. Retinitis pigmentosa was confirmed through ophthalmoscopy. Laboratory tests showed acanthocytosis, hypocholesterolemia, and markedly reduced lipid levels. ABL was diagnosed based on clinical presentation, laboratory findings, and a history of chronic diarrhea with fat malabsorption. Management included dietary modifications, vitamin supplementation, and diuretics, which initially improved the patient's condition. However, treatment complications such as Red Man syndrome and steroid-induced psychosis required "modifications to the management plan.
Conclusion: This case underscores the importance of early diagnosis and treatment of ABL, particularly in populations with a high prevalence of consanguinity. The patient's complex clinical trajectory highlights the challenges in managing ABL and the necessity for vigilant monitoring of treatment-related adverse effects. The case also emphasizes the value of genetic counselling and family screening in the management of inherited metabolic disorders.
Keywords: Abetalipoproteinemia; Bassen-Kornzweig Syndrome; Malabsorption Syndrome; Red Man Syndrome; Drug-Induced Psychosis.
Introduction
Abetalipoproteinemia (ABL) is a rare genetic" disorder marked by the lack of specific proteins and fat-soluble vitamins in the bloodstream. First documented in 1950, it is associated with abnormal red blood cells, retinal pigments, and ataxia. By 1960, researchers identified the absence of a particular protein in the blood of those affected. In 1992, it was discovered that individuals with ABL have reduced the activity of the microsomal triglyceride transfer protein (MTTP) in their intestines. Mutations in the MTTP gene were subsequently identified "in patients with ABL in 1993. The main symptoms of abetalipoproteinemia are primarily caused by the body's inability to absorb fat-soluble vitamins A, D, E, and K2. The main biochemical issue is the body's inability to properly absorb dietary fats and produce apolipoprotein (apo) B-containing lipoprotein particles. This is due to mutations in the MTTP gene, which encodes the microsomal triglyceride (TG) transfer protein large subunit (MTP). MTP is essential for assembling lipoproteins in the intestine and liver, where it transfers lipids to form chylomicrons and very low-density lipoproteins (VLDL). The lack of functional MTP results in extremely low levels of apo B-containing lipoproteins, total and LDL cholesterol, TG, and plasma biomarkers for vitamins A, D, E, and K in Abetalipoproteinemia patients [1].
Case Presentation:
We report on a 33-year-old Sudanese man," born to consanguineous parents, who has been experiencing progressive lower limbs swelling, facial puffiness, night blindness, and poor motor coordination. He has a history of recurrent fatty diarrhea, known as "steatorrhea," from childhood. Clinical examination revealed pale palms, Koilonychia, onycholysis, thin, brittle hair, and ophthalmoplegia. Ophthalmoscopic examination confirmed retinitis pigmentosa. Swelling was observed in his lower limbs extending to the knee joints, along with bruises on his knees. Given the clinical presentation, history of chronic diarrhea with fat malabsorption, and retinitis pigmentosa, an abetalipoproteinemia was considered a diagnosis along with other causes of fat malabsorption syndromes. Blood tests showed acanthocytosis Figure 1, low lipid levels, and hypocholesterolemia Table 1. The patient was treated with dietary modifications, vitamin supplementation, and diuretics. He showed improvement and was discharged after eight days with oral furosemide, metolazone, and spironolactone. Three months later, he experienced the same symptoms and was given furosemide injections along with vitamin supplements, metolazone, and spironolactone. He was anemic and received 4 units of blood, but developed a rare hypersensitivity reaction known as Red Man syndrome Figure 2. He was given dexamethasone to control the reaction, but unfortunately, the patient developed steroid-induced psychosis in the form of auditory hallucinations and sudden depressive episodes. Dexamethasone was stopped, and subsequently, the psychological manifestations subsided. An antihistamine injection was administered instead of dexamethasone, but the reaction persisted, so we decided to withdraw the medications one by one until we stopped the furosemide. He improved and was discharged after ten days of oral fat-soluble vitamins, metolazone, and spironolactone.
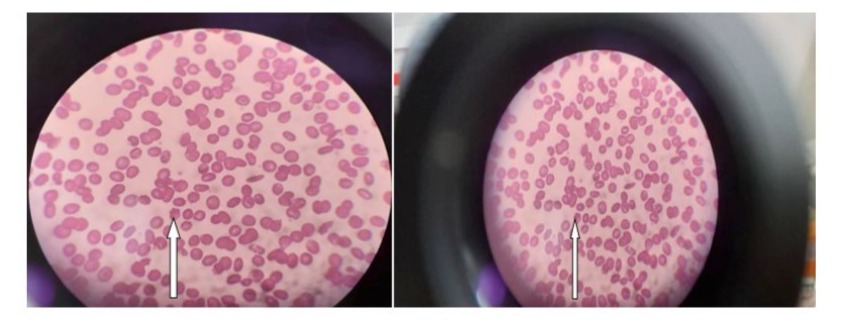
Figure 1: shows peripheral blood picture reveals Acanthocytosis.
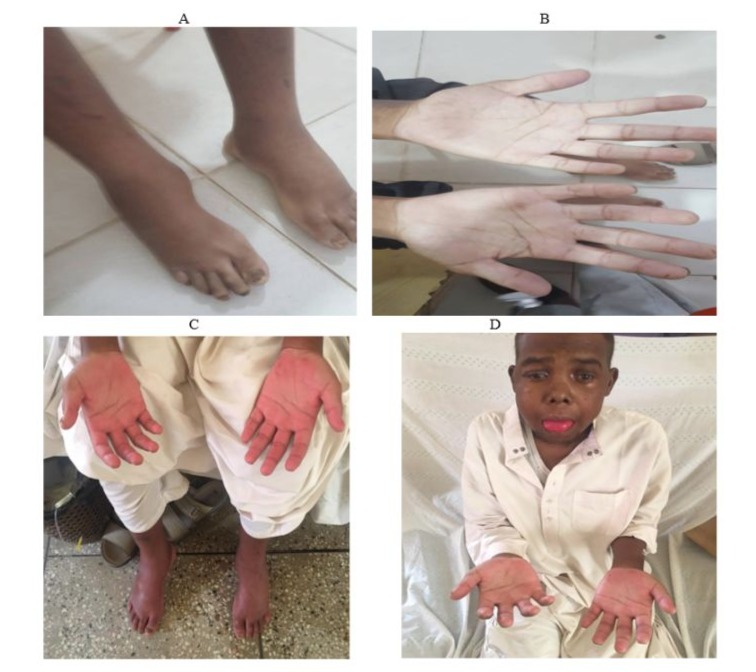
Figure 2: (A) showing lower limb edema, (B) showing pale palms, (C & D) showing red man syndrome.
|
Complete Blood Count |
Normal range |
Liver Function Test |
Normal range |
Renal Function Test and Electrolytes |
Normal range |
Bleeding profile |
Normal range |
||||
|
Hb |
5.3 |
12 – 16 g/dl |
T.Bilirubin |
3 mg/dl |
Up to 1 mg/dl |
Blood urea |
15ml/dl |
15 - 45 mg/dl |
PT |
25 |
12 - 17 sec |
|
RBCs |
1.57 |
3.8 – 5.0 mill cell/ cumm |
D.Billirubin |
0.7 mg/dl |
Up to 0.25 mg/d |
Serum Creatinine |
0.5mg/dl |
0.7 - 1.4 mg/dl |
INR |
1.7 |
Less than 1 |
|
TWBCs |
3.4 * 103 |
4 – 11 *103 cell/cumm |
Ind.Billirubin |
2.3 mg/dl |
Up to 0.75 mg/dl |
Serum Sodium |
138 mmol/dl |
135 – 145 mEq/l |
PTT |
43 |
20 - 45 sec |
|
Gran |
1.9*109 cell/cumm |
S.ALP |
124 u/l |
Up to 115 u/l |
Serum potassium |
2.7 mmol/dl |
3.5 – 5.3 mEq/l |
||||
|
Lymph |
1.1 * 10 9cell/cumm |
||||||||||
|
Plts |
86.000 |
150 – 450 * 103 cell/cumm |
|||||||||
Table 1: shows baseline investigations that reveal pancytopenia, indirect hyperbilirubinemia, hypokalemia, prolonged PT and INR, and hypocholestrolemiia.
|
Lipid profie |
Normal range |
|
|
Fasting S. chlestrool |
66 mg/dl |
120-200 mg/dl |
|
Fasting S. triglycerides |
94 mg/dl |
150-200 mg/dl |
Discussion
Clinical presentation of ABL is classical, as described in the literature, starting in childhood as failure to thrive with diarrhea and vomiting, fat malabsorption with steatorrhea, hepatomegaly, loss of night vision and/or color vision, and acquired atypical pigmentation of the retina. Progressive loss of deep tendon reflexes, vibration, proprioception sensation, ophthalmoplegia, slurred speech, and may be complicated with Friedrich's-like ataxia with broad base high stepping gait in early adulthood if remains untreated. Rarely, cardiomegaly and hypothyroidism were reported 1, 2, and 4. No official clinical diagnostic criteria exist for abetalipoproteinemia, so it is diagnosed based on the absence or extremely low levels of LDL-cholesterol, triglycerides, and apolipoprotein B, along with the identification of biallelic pathogenic variants in the MTTP gene through molecular genetic testing, which is the confirmatory diagnostic method [2,3]. Abnormally high levels of liver transaminases, prolonged international normalization ratio, and low levels of serum concentrations of fat-soluble vitamins (A, K, E, and D) can be detected. Additionally, intestinal endoscopic examination may reveal fatty deposition in the intestinal mucosa, which is known as snow-whit duodenum [2].
Management: Since ABL is a genetic disorder, treatment aims to manage the symptoms, signs, and complications through the following recommendations: Growth deficiency: Ensure sufficient caloric intake. Follow a low-fat diet (10%-20% of total calories), as a fat intake exceeding 20% is generally not well-tolerated. Oral supplementation with essential fatty acids, up to a teaspoon per day of oils rich in polyunsaturated fatty acids as tolerated. Steatotic liver without fibrosis: Restrict dietary fat. Since the fatty liver condition occurs without active inflammation, anti-inflammatory treatments are unnecessary. Liver transplantation will be considered in cases where hepatic fibrosis and/or cirrhosis have developed; administration of oral fat-soluble vitamins like vitamin A (100-400 IU/kg/day) to halt the progression of visual impairment and prevent eye complications; vitamin K (5–35 mg/week) to normalize abnormal INR; vitamin E (100-300 IU/kg/day) along with speech and language therapy will prevent and improve dysarthria; vitamin D (800–1200 IU/day); and occasionally, supplement with vitamin B12 and/or iron in case of mild anemia; and coordinated physiotherapy and intensive rehabilitation are necessary for controlling ataxia. Pregnancy management: Excessive vitamin A intake can pose risks to the developing fetus, so it is advised that pregnant women or those planning to conceive should cut their vitamin A supplement dosage by half. It is also recommended to closely monitor beta-carotene levels in the blood during pregnancy. Despite these precautions, it's important to note that vitamin A supplementation should not be stopped entirely during pregnancy as it is an essential nutrient [3]. Family counselling: Abetalipoproteinemia is passed down through autosomal recessive inheritance. When a child is conceived, each sibling of an affected person has a 25% probability of being affected, a 50% chance of being a carrier without symptoms, and a 25% chance of being unaffected and not a carrier. It is feasible to conduct carrier testing for at-risk family members and prenatal or pre-implantation genetic testing if the specific pathogenic MTTP gene variants in the family are identified [3].
Prognosis: Previous case studies have produced varied" findings on the potential connections between the age at which abetalipoproteinemia (ABL) is diagnosed, the initiation of a low-fat diet and vitamin replacement therapy, the specific mutation in the MTP gene, and the APOE genotype, and the long-term prognosis for ABL patients. In terms of prognosis based on age at diagnosis, ABL patients are commonly diagnosed in their second to fourth decades, with a few cases occurring in the first and sixth decades. An early diagnosis may indicate a more severe form of the condition and potentially a poorer prognosis, whereas later diagnosis may also result in a more severe outcome due to prolonged untreated vitamin deficiency, particularly during growth and development. Regarding the relationship between the type of MTP mutation and long-term prognosis, the limited available data makes it challenging to make predictions based on MTP genotype. Additionally, a recent meta-analysis revealed a linear relationship between APOE genotype and plasma LDL cholesterol levels. Some mutations in the MTP gene have been suggested to significantly reduce LDL cholesterol levels, particularly in individuals with APOE E2 homozygosity. A study involving six ABL patients suggested that the E4/E2 genotype may be associated with less favorable outcomes [4-6].
Authors Contribution Statement
MIA consultant physician was involved in making the decisions regarding the patient's treatment while in the intensive care unit. He was the primary consultant physician of the patient. He reviewed the manuscript critically and approved it for final submission. IBM, MML, and IBA were the internship doctors of medicine whom took care of the patient. They were involved in planning of various investigations for the patient and performed the literature review, drafted the manuscript, reviewed it critically and approved it the end for submission. They were also involved in retrieving high quality images for publication from the haematology and radiology archives. All authors read and approved the final manuscript.
Conflict of Interest:Conflict of interest declared none.
Ethical Considerations: Informed consent from patient.
Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- Vlasschaert C, McIntyre AD, Thomson LA, Kennedy BA, Ratko S, et al (2021) Abetalipoproteinemia Due to a Novel Splicing Variant in MTTP in 3 Siblings. J Investig Med High Impact Case Rep. 9:23247096211022484.
- Takahashi M, Okazaki H, Ohashi K, Ogura M, Ishibashi S, et al (2021) Current Diagnosis and Management of Abetalipoproteinemia. J Atherosclerosis Thrombi. 28:1009-1019.
- Hegele RA, Hooper AJ, and Burnett JR. (2018) Lipoproteinemia abeta. 2018 Oct 25 [updated 2022 May 19]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024.
- Gurram S, Holla VV, Sriram N, Phulpagar P, Jha S, et al (2022) A Rare Case of Ophthalmoplegia with Ataxia in Genetically Proven Abetalipoproteinemia. Mov Disord Clin Pract. 10:514-517.
- Rodríguez Gutiérrez PG, González García JR, Castillo De León YA, Zárate Guerrero JR, Magaña Torres MT. (2021) A novel p.Gly417Valfs*12 mutation in the MTTP gene causing abetalipoproteinemia: Presentation of the first patient in Mexico and analysis of the previously reported cases. J Clin Lab Anal. 35:e23672.
- Zamel R, Khan R, Pollex RL, Hegele RA. (2008) Abetalipoproteinemia: two case reports and a literature review. Orphanet J Rare Dis. 3:19.
© by the Authors & Gavin Publishers. This is an Open Access Journal Article Published Under Attribution-Share Alike CC BY-SA: Creative Commons Attribution-Share Alike 4.0 International License. Read More About Open Access Policy.